He pensado en tomarme unos minutos para hablar sobre este tema, ya que es muy frecuente que discutamos sobre el tema central, la validación de las planillas Excel, pero siempre que tenemos la oportunidad, en talleres, en cursos o simplemente cuando hacemos una evaluación de riesgo para saber como estamos en este tema, surge la pregunta…
¿Cómo administramos las planillas Excel?, ¿Lo hacemos de una forma segura?
Veamos un poco el tema.
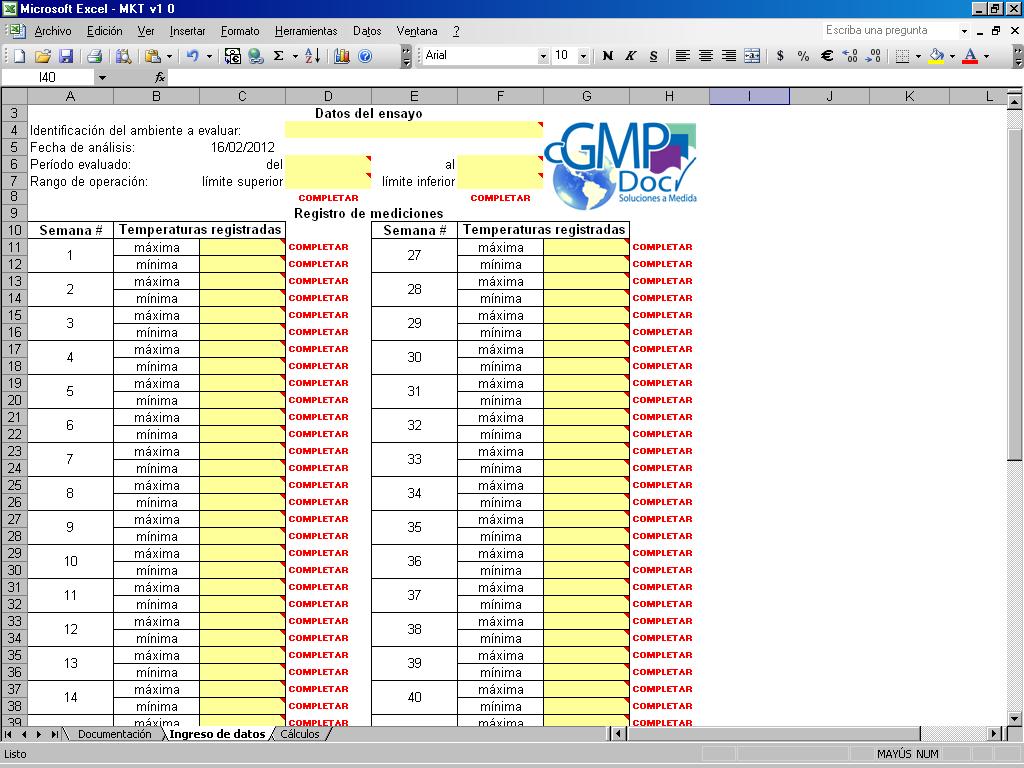
Les recuerdo que hemos hablamos de la importancia de determinar el impacto GMP de una planilla Excel (PE), considerando, si la misma tiene impacto directo sobre la pureza, seguridad, eficacia, identidad del producto, o la medición o monitoreo de estos atributos, o si el Excel produce datos que serán usados para aceptar o rechazar el producto, o también si los resultados obtenidos del Excel interactúan con otros sistemas que tienen efecto directo sobre la calidad del producto.
También mencionamos la construcción de las planillas y las actividades de validación para asegurar que las mismas, fórmulas, instrucciones, macros, etc., cumplan con las especificaciones y sean seguras.
Pero hoy quiero hacer foco en dos aspectos muy importantes: la administración de las planillas y el uso apropiado de las mismas.
Pero que pasa luego que la planilla ya está validada y el personal del laboratorio está entrenado en su uso?
El responsable de administrar las planillas del laboratorio (usualmente QA), debe proteger las planillas, usualmente asignarles una codificación, podría ser por ejemplo:
PE-NNN vXX
Donde:
PE: acrónimo de Planilla de cálculo Excel
NNNN: número único para cada PE, comenzando por el 001
V: correspondiente a versión
XX: número de
Luego QA registra la información de la PE en un listado o inventario de planillas Excel del laboratorio conteniendo las planillas validadas vigentes.
Desprotege la PE y workbook.
Asegura que cada impresión del worksheet incluye la siguiente información en cada página:
- Nombre del archivo / nombre del worksheet (hoja) en el encabezado
- Número de página en el pié de página
- Estado y fecha de vigencia en el pié de página
- Ruta donde está la planilla de cálculo Excel en el pié de página
- Fecha y hora de impresión
Verifica que la información agregada no altera el número de páginas impresas (ajusta de forma necesaria para mantener el número de páginas como está testeada y documentada en los test del usuario de la PE).
Respeta los códigos de color establecidos para la fuente y las celdas.
Setea la PE para ser compartida, de la siguiente manera, accede al ítem menú: Tools>share workbook y chequea el ítem “permitir cambios por más de un usuario al mismo tiempo”.
Protege la PE y el workbook con un password. Ingresa al menú Herramientas / proteger Hoja y hace click, ingresa la contraseña y luego presiona “enter” o click en el botón aceptar. Registra el password (s) en el inventario de planillas validadas vigentes. Salva el archivo y lo cierra. Coloca la PE validada en la carpeta de la red dedicado para ese propósito y liberada para su uso.
Para efectuar la administración de las Planillas Excel, suelen crearse carpetas para ubicar a las planillas según su status, por ejemplo:
- Planillas Validadas y vigentes
- Planillas No vigentes
- Planillas en modificación / validación
- Templates o modelos (como por ej. Especificación de diseño, protocolo de validación, etc.)
Los permisos de acceso a tales carpetas son los siguientes:
| Posición | Validadas/vigentes | No vigentes | En modificación/validación |
| Supervisor QC | Solo lectura | No acceso | Copia/borra/modifica |
| Usuario | Solo lectura | No acceso | No acceso |
| Personal QA | Acceso completo | Acceso completo | Acceso completo |
| Adm. IT | Backup / recovery | Backup / recovery | Backup / recovery |
Uso de las planillas Excel validadas:
Cada usuario debe verificar que la planilla está disponible en la carpeta de planillas de cálculo Excel vigentes.
Cada impresión de planilla debe incluir los nombres del archivo y planilla, la información del número de página, la fecha y la hora de impresión, el estado de la planilla y la fecha de vigencia.
En cuanto al uso de las mismas, quiero que recuerden que dado que las planillas Excel “perse” no cumplen con la parte 11, el uso esperado es abrir la planilla, completar los campos necesarios con datos, generar una impresión, revisarla y firmarla.
No está permitido que los usuarios finales salven la PE de manera de permitir que el archivo sea cargado de la misma forma cada vez que se accede a él.
Cambios a una PE existente, los mismos deben ser efectuados a través del sistema de control de cambios o a través de un log de cambios de la planilla, de manera que las modificaciones sean documentadas y aprobadas antes de ser efectuadas.
Una vez aprobado el cambio, QA efectúa una copia de la planilla existente para que sea efectuada la nueva versión, luego de creada la misma sigue el mismo camino que vimos anteriormente.
La planilla modificada reemplaza a la anterior, la cual es retirada a obsoleta.
Espero que les resulte útil.


 Bueno, pero afortunadamente en la actualidad hay disponibles sistemas de gestión de documentos o DMS (Document Management System).
Bueno, pero afortunadamente en la actualidad hay disponibles sistemas de gestión de documentos o DMS (Document Management System).