Si Ud. quiere mejorar tiene que hacer algo diferente.
A continuación le dejamos un cuestionario sobre 10 temas GMP:
Políticas y Procedimientos:
- Sus procedimientos son escritos por los usuarios y para los usuarios?
- Están disponibles en los lugares de trabajo?
- Sus políticas describen el por qué (visión general) y sus procedimientos el cómo (detalles)?
- Utiliza figuras, esquemas y flujogramas para darles más claridad?
- Tienen el nivel de detalle apropiado para el usuario?
- Ensaya los procedimientos claves antes de implementarlos?
- Revisa, corrige y mejora los procedimientos claves después de 6 meses de uso?
Educación:
- Hace foco en la educación (cambios de conducta) NO solo en cumplir con el entrenamiento?
- Tiene una estrategia de educación a 3-5 años?
- Dispone de un presupuesto para educación?
- Sus programas de educación están diseñados para satisfacer todos los estilos de aprendizaje?
- La mayoría de su educación ocurre en el lugar de trabajo, o sea fuera de la sala de capacitación?
- evita que el entrenamiento termine en la presentación?
- Hace foco en proveer el fundamento básico (el porqué)?
- El repaso de las GMP, se renueva anualmente?
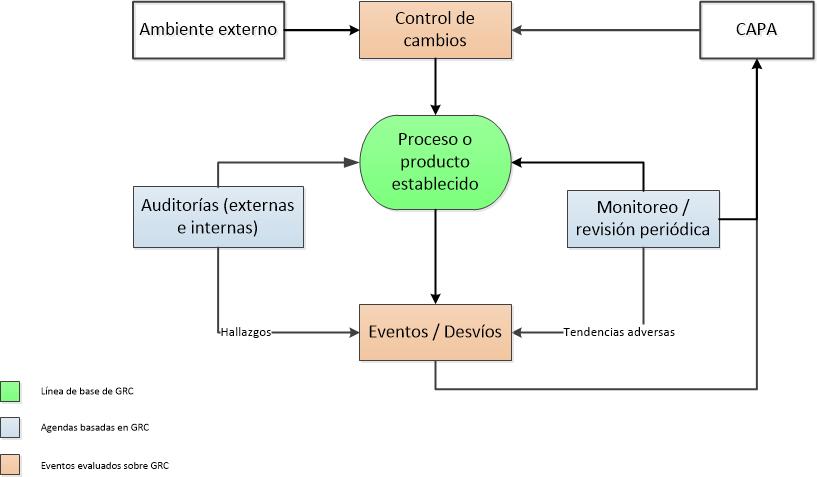
Gestión de Riesgos de Calidad (GRC):
- Aplica la GRC para la priorización y la mejora continua y NO para apagar incendios, manejar las crisis o cuando las cosas salen mal?
- Acepta que no existe el riesgo cero?
- Los principios y las prácticas de la GRC son entendidas a todos los niveles (desde el Gerente de la Planta hasta los operadores de la misma)?
- Usa la GRC a lo largo del ciclo de vida del producto? Por ejemplo:
- Para enfocar sus actividades de validación?
- Para escalar los eventos de calidad?
- Para optimizar sus recursos de auditorías?
- Para clasificar los desvíos o reclamos de clientes?
- Para optimizar el monitoreo ambiental?
- Para proporcionar un marco para la toma de decisiones basado en el riesgo?
- Hay un excelente conocimiento de sus procesos y productos a lo largo de su empresa… sin el cual es imposible la GRC?
- NUNCA usa la GRC para justificar lo que sabe que es de baja calidad?
Manejo de desvíos y sistema CAPA:
- Tiene una cultura abierta, libre de culpables, donde las personas son alentadas a reportar los desvíos?
- Los incidentes son reportados inmediatamente, sin demoras?
- Efectúa una clasificación de los incidentes objetivamente dentro de 24 hs. Para priorizar los recursos y efectuar las investigaciones?
- Inicia sus investigaciones dentro de un día de trabajo hábil, como máximo?
- Investiga proporcionalmente al riesgo, en vez de tratar cada incidente de la misma forma?
- Las investigaciones son efectuadas donde ocurre el incidente, o son manejadas detrás de un escritorio?
- Las investigaciones son hechas por personas con conocimiento de los procesos y productos, o sólo por QA?
- Los CAPAs están focalizados en prevenir la re-ocurrencia en vez de ocuparse de los síntomas superficiales?
- Implementa CAPAs tan pronto como es posible, o cae en asignarle por regla 30 días?
- Cree que el error humano es la “consecuencia” y rara vez la “causa” de los desvíos?
- Lleva una tendencia de los eventos repetidos?
- Comparte incidentes y CAPAs a lo largo de la organización para fomentar la mejora continua?
- Arma una agenda para verificar la efectividad de cada CAPA para evaluar cuan exitosos han sido para prevenir la re-ocurrencia del desvío?
Métricas de Calidad y KPI (Indicadores Claves de Performance):
- Tiene métricas orientadas a la mejora de los procesos?
- Usa usted el enfoque de “Pareto”, centrándose en el 20% de las métricas que proporcionan el 80% del beneficio?
- Son sus métricas de “propiedad” de los usuarios? Las mismas se ven impulsadas hacia arriba, no de arriba hacia abajo?
- Los datos son compartidos con los usuarios y los grupos de interés para impulsar la mejora continua?
Auditorías internas y autoinspecciones:
- Su programa está basado en el riesgo?
- Tiene un proceso de escalada rápido y efectivo para las observaciones críticas y las tendencias negativas?
- Sus auditores están plenamente calificados, educados a partir de todas las áreas, no sólo de aseguramiento de calidad?
- Sus auditores ofrecen consejos prácticos y soluciones, no solo críticas?
- Las inspecciones son usadas para agregar valor, no solo por el bien de compliance?
- Para ayudar a prevenir problemas
- Para compartir las mejores prácticas en todas las áreas
- Para expulsar a la complejidad, no crearla
- Hace una tendencia de los hallazgos de la auditoría para evaluar la imagen completa?
- Sus programas de auditorías internas y autoinspecciones aseguran que está listo para ser inspeccionado en todo momento?
Sistema de control de cambios:
- Su sistema de control de cambios ha sido diseñado con la participación activa de todas las partes interesadas y no solo QA?
- Su sistema de control de cambios es simple y entendible para todos?
- El control de los cambios es considerado vital para la mejora del negocio y la gestión de los riesgos o solo se lo considera por un tema de cumplimiento de las regulaciones?
- El sistema tiene un requerimiento del cambio el cual es revisado y aprobado en hs o días, no meses?
- Su sistema de control de cambios utiliza una evaluación de impacto basada en los riesgos como soporte para la aprobación o el rechazo de las solicitudes de cambios?
- Efectúa el seguimiento de todos los cambios aprobados para asegurarse que los mismos fueron efectivos?
Validación:
- Sus actividades de validación están bajo un Plan?
- Dispone de los recursos necesarios para el cumplimiento de las actividades de validación?
- Realmente entiende la variabilidad el proceso?
- Cada uno entiende o conoce los puntos críticos de control del proceso?
- Cada uno entiende los atributos claves del producto, aquellos que impactan en la seguridad del paciente?
- Sus métricas confirman un proceso bajo control? “Cero” retrabajos y/o reprocesos, 100% correcto a la primera vez?
Integridad de datos:
- Todo su personal entiende que una pobre integridad de los datos, genera pérdida de confianza regulatoria?
- Tiene back up y archivo de sus datos?
- Rutinariamente revisa y comprueba la integridad de los datos?
- Rutinariamente desafía todas las transacciones (audit trail)?
- Tiene validados sus sistemas de gestión de datos?
Su Cultura de Calidad:
- Qué hace la gente cuando nadie la observa?
- QA está integrado a la planta?
- Los supervisores dedican más tiempo a la planta que a las reuniones?
- Cada uno tiene objetivos personales de performance de calidad?
- Sus líderes predican con el ejemplo y demuestran las conductas de calidad correctas?
- La gente habla sobre Calidad, no solo sobre resultados?
- Desarrolla, recompensa y retiene los conocimientos en lugar de permitir que dejen la organización cada vez que vean algo mejor?
- Trata a sus proveedores como una extensión de su línea de producción?
- Selecciona a sus proveedores en base a calidad, no en base a precio?
- Cada uno se siente conectado al paciente?
- Regularmente compara sus estándares con los del mejor en su categoría?
Si alguna de las respuestas no fue del todo satisfactoria, Ud. ha detectado una oportunidad de mejora sobre alguno de los 10 temas, asegúrese tener un plan de acciones de remediación.
Desde cGMPdoc podemos darle soporte en entrenamientos, implementación o actualización de procesos, auditorías, evaluación de sus proveedores, consulte en info@cgmpdoc.com