Posts tagged ‘desvíos’
Les dejo algunas preguntas para autoevaluarse respecto de manejo de la GRC:
Ud aplica la GRC para la priorización y la mejora continua y NO para apagar incendios, manejar las crisis o cuando las cosas salen mal?
Ud. Acepta que no existe el riesgo cero?
Todo el personal tiene un claro entendimiento del umbral de riesgo de la empresa?
Los principios y las prácticas de la GRC son entendidas a todos los niveles (desde el Gerente de la Planta hasta los operadores de la misma)?
Ud. usa la GRC a lo largo del ciclo de vida del producto? Por ejemplo:
- Para enfocar sus actividades de validación?
- Para escalar los eventos de calidad?
- Para optimizar sus recursos de auditorías?
- Para clasificar los desvíos o reclamos de clientes?
- Para optimizar el monitoreo ambiental?
- Para proporcionar un marco para la toma de decisiones basado en el riesgo?
Hay un excelente conocimiento de sus procesos y productos a lo largo de su empresa… sin el cual es imposible la GRC?
NUNCA usa la GRC para justificar lo que sabe que es de baja calidad?
Si alguna de las respuestas no fue del todo satisfactoria, Ud. ha detectado una oportunidad de mejora sobre la Gestión de riesgo de calidad, asegúrese tener un plan de acciones de remediación, también desde cGMPdoc podemos proponerle una solución o le dejamos a mano la posibilidad de un taller “In Company” teórico-práctico, consulte en info@cgmpdoc.com
{kind=link}
{kind=link}
Les dejo 13 preguntas para autoevaluarse respecto de manejo de desvíos y sistema CAPA:
- Tiene una cultura abierta, libre de culpables, donde las personas son alentadas a reportar los desvíos?
- Los incidentes son reportados inmediatamente, sin demoras?
- Efectúa una clasificación de los incidentes objetivamente dentro de 24 hs. Para priorizar los recursos y efectuar las investigaciones?
- Inicia sus investigaciones dentro de un día de trabajo hábil, como máximo?
- Investiga proporcionalmente al riesgo, en vez de tratar cada incidente de la misma forma?
- Las investigaciones son efectuadas donde ocurre el incidente, o son manejadas detrás de un escritorio? Siempre?
- Las investigaciones son hechas por personas con “know how” de los procesos y productos, o sólo por QA?
- Los CAPAs están focalizados en prevenir la re-ocurrencia en vez de ocuparse de los síntomas superficiales?
- Implementa CAPAs tan pronto como es posible, en vez de aplicar la “infundada regla de los 30 días”?
- Cree que el error humano es la “consecuencia” y rara vez la “causa” de los desvíos?
- Lleva una tendencia de los eventos repetidos?
- Comparte incidentes y CAPAs a lo largo de la organización para fomentar la mejora continua?
- Arma una agenda para verificar la efectividad de cada CAPA para evaluar cuan exitosos han sido para prevenir la re-ocurrencia del desvío?
Si alguna de las respuestas no fue del todo satisfactoria, Ud. ha detectado una oportunidad de mejora sobre el manejo de los desvíos y sistema CAPA, asegúrese tener un plan de acciones de remediación, también desde cGMPdoc podemos proponerle una solución o le dejamos a mano la posibilidad de un taller “In Company” teórico-práctico.
Les dejo una lista de 10 ítems a considerar a la hora de calificar un contratista o tercero:
1. Existencia de QAA (Quality Assurance Agreement) disponible, aprobado y en cumplimiento.
2. Sistemas de Calidad, in place y efectivos.
3. Robustez del proceso de investigación del tercero o contratista.
4. Nivel de comunicación con el laboratorio, ante inspecciones recibidas por ellos o antes eventos que pudieran suceder.
5. Perfil GMP
6. Acciones frente al mercado, por ej. freno de operaciones ante un Recall.
7. Manejo de los cambios
8. Desvíos significativos, tasa de ocurrencia, disponibilidad de los reportes requeridos para la liberación del lote.
9. Reclamos de calidad, cuan efectivo es el sistema de manejo de quejas.
10. Historia de los riesgos, hay riesgos capturados?
Uno podría establecer respecto de estos 10 ítems una clasificación de la siguiente manera:
Riesgo alto: 2 o + de estos ítems altos
Riesgo medio: 1 ítem alto o 2 o + medios
Riesgo bajo: 1 ítem medio y otro bajo.
Bueno esta es una forma de poder evaluar el nivel de riesgo de mi contratista o tercero. Esta claro que debemos efectuar una revisión de todos estos ítem y ponderarlos cuali o cuantitativamente, de manera de poder calificarlos.
Espero que les resulte útil.
Hay dos componentes necesarios para que una auditoría sea exitosa.
El primero es un auditor con las habilidades, la educación y la experiencia correcta o necesaria (ver haciendo click aquí).
El segundo es el proceso de auditoría por sí mismo.

El proceso de auditoría puede comenzar algunos meses antes de que la auditoría se lleve a cabo. Las etapas del proceso de auditoría son:
Preparación de la auditoría
- Determinar quién conducirá la auditoría – un individuo o un equipo.
En el caso de un equipo, el auditor líder será responsable de asegurar que la auditoría es manejada efectivamente y eficientemente.
- Desarrollar la agenda de la auditoría
Para conducir una auditoría eficiente y efectiva, debe ser elaborada una agenda. Esto ayudará a mantener el flujo de la misma sin problemas y hará mejor el tiempo del auditado y del auditor.
El propósito de la auditoría debería estar claro para ambos, el auditado y el auditor. Puesto que efectuará una auditoría GMP, Ud. Debería considerar los siguientes temas GMP:
- Sistemas de Calidad
- Gestión de materiales
- Controles de calidad del laboratorio
- Sistemas de producción
- Instalaciones y equipos
- Empaque y rotulado
Durante la auditoría el auditor debe planear prestar especial atención a los sistemas de calidad, la forma en la cual el laboratorio maneja los desvíos, los reclamos y los resultados de los ensayos OOS (fuera de especificaciones), esto le dará una visión de la cultura de calidad de la compañía y su adherencia a las cGMPs.
- Confirmar la auditoría y acordar una fecha con el proveedor /área a ser auditada.
Trabajar con el auditado para determinar un tiempo para ajustarse dentro del marco de tiempo que sea conveniente para ambas partes.
- Revisar documentos, incluir documentos recibidos desde la empresa o el laboratorio
- Preparar notas para la auditoría
Luego de revisar la información de soporte y los documentos de referencia y los estándares, el auditor prepara notas que lo ayudará a efectuar la auditoría, así como una lista de documentos a revisar.
Potenciales actividades de la auditoría en el laboratorio
Todas las auditorías deben ser conducidas de una forma abierta, el auditor debe discutir los hallazgos y expectativas con el auditado mientras conduce la auditoría. La auditoría es una oportunidad de identificar oportunidades de mejora para el auditado.
- Conducir una reunión de apertura
Al arribar al laboratorio, el auditor / equipo auditor debe efectuar una reunión de apertura con los responsables del laboratorio (QA, producción, dirección, otros representantes asignados). Durante la reunión de apertura, el auditor debe asegurarse que el auditado entienda su rol en la auditoría.
- Recorrer los depósitos, instalaciones de manufactura y laboratorios, si es apropiado.
Las auditorías pueden comenzar con una recorrida a las instalaciones y al laboratorio, es recomendable “seguir un producto” (desde la recepción de materiales hasta la liberación del producto terminado).
Durante la recorrida, verificar que los SOPs e instrucciones están siendo seguidos y los registros se completan con exactitud.
A medida que realiza la auditoría el auditor puede seguir las notas preparatorias, para asegurar que cubre la auditoría completa.
Las observaciones de la auditoría pueden ser clasificadas en críticas, mayores y menores.
Si un tema o desvío crítico es observado, el mismo debe ser comunicado inmediatamente al management del laboratorio y al responsable auditado
- Entrevistar analistas y operadores (si es necesario).
- Efectuar una reunión informativa de la auditoría con el auditado, si es apropiado.
Para las revisiones informativas donde las observaciones y hallazgos deberían ser revisadas con el auditado como también las áreas de preocupación para asegurar que no habrá sorpresas o malos entendidos en el cierre final de la auditoría.
La agenda de la auditoría puede ser actualizada si son requeridos cambios basados en las observaciones o recursos.
- Efectuar reuniones de auditoría con el equipo, si un enfoque de equipo auditor es utilizado.
Estas reuniones son mantenidas para ayudar al equipo auditor a mantenerse enfocado en el objetivo de la auditoría y la auditoría en curso.
- Revisar la documentación del auditado
Esto puede incluir SOPs, documentación de manufactura y de ensayos, datos crudos asociados, protocolos de calificación y validación, y reportes de investigaciones.
- Conducir la reunión de cierre
La reunión de cierre es el foro usado para presentar verbalmente los resultados de la auditoría al management auditado. Es esencial que todos los participantes claves de la auditoría y el management del auditado participen de la reunión de cierre.
Seguimiento de las actividades de la auditoría
- Documentar las observaciones en un reporte formal
Los mismos están basados en las anotaciones que el auditor tomó durante la auditoría. Los mismos pueden ser usados por diferentes personas para diferentes motivos, por eso necesitan ser claros y escritos de una forma consistente como también contener la información relevante y necesaria.
Los reportes de auditoría deben ser normalmente emitidos dentro de un cierto plazo, así como también las respuestas conteniendo el plan de acción.
- Evaluar las respuestas recibidas desde la empresa / laboratorio
Luego que las respuestas son recibidas, es necesario analizarlas y determinar si las acciones correctivas planteadas y los riesgos propuestos son aceptables. Una vez recibida la respuesta, y si las respuestas son consideradas aceptables, el auditor tiene la responsabilidad de responder dentro de un período de tiempo definido.
- Cerrar la auditoría
- Efectuar el seguimiento de los CAPAs generados
El seguimiento puede requerir nuevas visitas al laboratorio o la recepción de las evidencias de las acciones cumplidas, para darle cierre a la acción pendiente.
Evaluación de riesgo durante el proceso de auditoría:
El auditor toma decisiones basadas en el riesgo a través del proceso de auditorías, por ejemplo dado que No es posible cubrir todo durante el proceso de auditoría, es necesario evaluar los riesgos, tal vez efectuar un análisis de riesgo formal y hacer un juicio sobre que cubrir. Este juicio que necesita hacer puede necesitar cambiar durante el proceso de auditoría basado en nuevos conocimientos.
Herramientas de análisis de riesgos:
Para soportar la evaluación de riesgo de un proveedor, el auditor debe elegir usar una herramienta de análisis de riesgo. Una de las herramientas formales de análisis de riesgo usadas por los auditores es Failure Modes Effects Analysis (FMEA).
Si Ud. está interesado en entrenar a su equipo auditor, consulte en info@cgmpdoc.com. Podemos ofrecerle materiales de entrenamiento para Auditores, check lists para que Ud. Efectúe las distintas auditorías y además tenemos la posibilidad de entrenar a su equipo “In Company”.
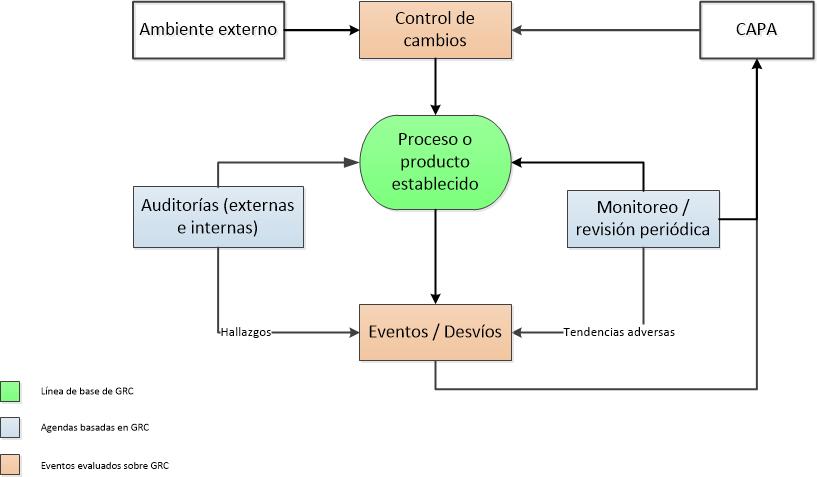
Hoy quiero referirme a un sistema de calidad, el sistema CAPA, sin embargo antes de comenzar a hablar específicamente sobre dicho sistema, quiero referirme al enfoque basado en el riesgo que están adoptando las agencias regulatorias y mencionar algunas preguntas que nos hacemos cuando intentamos comprender este tema, como por ejemplo: • ¿Cuál es el riesgo y para quién? • ¿Qué eventos causan o incrementan el nivel de riesgo? • ¿Cómo se manifiesta el riesgo? • ¿Cómo definir los niveles de riesgo? Pero antes veamos como la ICH Q9 define el riesgo:
“La combinación de la probabilidad de ocurrencia de daño y la gravedad de dicho daño. “Por lo tanto, el riesgo es asociado con un daño detectable, que puede ser medido a través de una probabilidad y severidad.
En el proceso de fabricación de productos, el riesgo se asocia con un evento que pueda comprometer la calidad, seguridad, y / o la eficacia del mismo. El medicamento comprometido podría dañar a los pacientes y al público en general, también en algunos casos, el riesgo podría afectar el personal de la fabricación de la droga, o a la empresa en sí, por ejemplo, ya que si la misma no cumple los requerimientos regulatorios puede ser multada o inhabilitada.
Centrándonos en el riesgo al paciente, es obligación del fabricante de medicamentos reducir la probabilidad de ocurrencia y minimizar la gravedad de los daños cuando estos eventos ocurren. Todas las actividades humanas y los emprendimientos tienen riesgo asociado con ellos. La elaboración de los medicamentos NO es la excepción.
Los eventos que suceden durante la elaboración de los medicamentos deben ser revisados y su riesgo evaluado.
Dependiendo del nivel del riesgo, la importancia o profundidad del análisis y la toma de acciones. Consideramos como de alto riesgo aquello que impacta directamente en el producto.
¿Cómo se manifiesta el Riesgo? Desde un punto de vista GMP, el riesgo depende del peligro que se plantea para el paciente cuando los eventos que suceden no son detectados y tratados adecuadamente antes de la la distribución del producto. Por ejemplo pueden ser liberados al mercado productos contaminados o con una potencia menor de la declarada.
Es sumamente importante ser consientes de las formas en que los riesgos se manifiestan, para poder tener un alto nivel de detectabilidad y tomar las acciones correctivas o preventivas apropiadas. Estos son componentes importantes para la estrategia de reducción de riesgos.
En orden de desarrollar la CAPA adecuada para mitigar el riesgo, uno tiene que definir y priorizar los niveles de riesgo.
Los niveles de riesgo y el Número Probabilidad de Riesgo (RPN) En la fabricación de productos farmacéuticos, el nivel de riesgo para una evento de calidad puede ser identificado a través de la combinación de la Severidad del daño al paciente, la frecuencia por la que la evento ocurre, y la detectabilidad del evento. Estos tres factores generalmente se les asigna un valor numérico y la multiplicación de los mismos da lugar al RPN= número de probabilidad de riesgo.
La severidad de un evento de calidad dado es una medida de la consecuencia del suceso en sí mismo y su potencial daño a la paciente, por ejemplo los valores más altos se lea signan a lesiones muy graves o muerte del paciente y los valores más bajos a eventos que no causan molestias. La frecuencia de un evento dado define la probabilidad de su aparición / reaparición. Los valores más altos son para una certeza que el evento ocurre u ocurrirá a situaciones que no pasan, quizás pasaron en el pasado, pero no ahora. La detectabilidad es una medida de la probabilidad de que la calidad evento se detecta o su efecto / resultado será fácilmente medido o visto. Aquí, los eventos que no son detectables tienen el índice de detectabilidad más alto, mientras que los eventos fácilmente detectables tienen el índice más bajo de detectabilidad.
Una vez que se determina el nivel de riesgo de un evento de calidad, es necesario aplicar los principios de Q9, donde indica que los esfuerzos de investigación y las acciones tomadas deben ser adecuados con la magnitud del problema y proporcionales a los riesgos encontrados.
O sea que los eventos de alto riesgo requieren una investigación profunda, análisis de causa raíz, para luego tomar las acciones correctivas / preventivas apropiadas, ya que este evento NO debería volver a ocurrir.
CAPA (acciones correctivas y acciones preventivas) es un sistema de calidad diseñado para mitigar el riesgo en la elaboración de productos.
La ICHQ10 sugiere que las empresas farmacéuticas “Deben tener un sistema parala aplicación deacciones correctivas y preventivas… un enfoque estructurado paraelproceso de investigacióndebe ser utilizado conel objetivo dedeterminar la causa raíz”.
Como un sistema de calidad de reducción del riesgo, el sistema CAPA aborda eventos de calidad, que se producen durante el proceso de elaboración de productos para la salud. Como por ejemplo, desvíos o resultados OOS (fuera de especificación), etc.:
Estos eventos de calidad tienen el potencial de presentar riesgos para la población, de ahí la necesidad de mitigar su efecto.
Y como no podría ser de otra forma, el evento debe ser documentado, su investigación, las conclusiones, las acciones que deben adoptarse y el calendario para su aplicación, el cierre de la desviación o no conformidad y la verificación de la efectividad de la misma.
El seguimiento manual representa retos asociados con la generación de demasiado papel, siendo engorroso y consume mucho tiempo, además de proporcionar acceso limitado. El seguimiento electrónico se está convirtiendo en una alternativa cada vez más utilizada, ya que elimina muchas de las deficiencias del seguimiento de manual. Sin embargo, estos sistemas de rastreo electrónicos agregan el requisito de ser 21 CFR parte 11 compliance, ya que deben generar y mantener los registros electrónicos.
Las etapas del Sistema CAPA:

Hasta este punto, la discusión se ha centrado en el riesgo en cumplimiento y de una forma a grandes rasgos, el enfoque general de un programa de CAPA. Ahora veamos un poco más de cerca el sistema CAPA.
Como se mencionó anteriormente, CAPA es un sistema de aseguramiento de la calidad, que se ocupa de eventos de calidad, que se puede producir o podría ser previsto que se produzca durante la fabricación de productos para la salud. El sistema se basa en la revisión del evento y analizar el riesgo asociado con el mismo. Dependiendo del RPN, la necesidad de generar acciones de mitigación. Una vez tomada la decisión, entonces se toma la acción apropiada. El evento, el análisis, las decisiones tomadas, y la acción (s) tomada luego son documentados, comunicados y seguimiento a asegurarse de que eran correctos, adecuados, y no lo hicieron introducir variabilidad riesgo / diferente o adicional a la operación. El proceso en 10 pasos básicos:
- Evento
- Identificar y segregar el producto afectado (colocarlo ON HOLD)
- Identificar si hay un equipo involucrado (colocarlo ON HOLD)
- Documentar, iniciar el desvío y documentar las acciones inmediatas
- Investigar y evaluar el riesgo
- Identificación de la causa raíz
- Toma de las acciones correctivas necesarias para disminuir o eliminar el riesgo (asegurar que ningún cambio adicional introduce un nuevo riesgo)
- Registrar y comunicar
- Monitorear el proceso de ejecución
- Verificar la eficacia de la acción tomada
Resumen
Los eventos de calidad, que se producen durante la fabricación de productos de salud, siempre están asociados con un nivel de riesgo. Un programa CAPA robusto es un requisito reglamentario que define el nivel de riesgo y cómo mitigarlo. Sin embargo, debe tenerse en cuenta que la implementación de un programa de este tipo no solo se limita a cumplir con la norma, sino también favorece al negocio y el aspecto financiero de la compañía. . Por otra parte, la implementación de un programa de CAPA no sólo tiene un impacto económico positivo en la fabricación de tales procesos, sino que también daría lugar a una mejor satisfacción del cliente y reduce el riesgo para el público, el principal objetivo.
Ya en otro artículo hemos definido desvío o no conformidad, voy a mencionar otra forma de definirlo:
Un evento no planeado que puede provocar el alejamiento de un proceso, procedimiento, resultado inesperado, no cumplimiento de un requisito previamente establecido en las cGMP.
También mencionamos que los podemos clasificar en críticos, mayores y menores de acuerdo a su impacto o gravedad.
Ahora no voy a volver sobre el proceso de investigación de los desvíos sino que quiero que veamos el tema de la eficacia de las investigaciones.
Si bien muchos utilizan la evaluación de la eficacia de los CAPAs, tomándose un tiempo posterior a la implementación del CAPA y revisando los resultados, hoy quiero referirme a la identificación de los desvíos repetidos.
La evaluación del desvío para ver su reincidencia, se hace sobre un período de tiempo (puede ser 12 – 24 meses).
Un desvío repetido es un evento que se repite con la misma causa raíz de cualquier clase de desvío para el mismo o similar producto / material, proceso, equipo, área, documentación u otro sistema GMP dentro del período de tiempo establecido (12-24 meses).
Pero cual es la interpretación de este hecho?:
La ocurrencia de un desvío repetido indicaría que la investigación inicial del desvío no parecería haber sido adecuada en la identificación de la causa raíz del evento o no se completó adecuadamente la acción correctiva para prevenir su reocurrencia.
Para los interesados, les dejo un flujograma muy simple para la identificación de los desvíos repetidos:
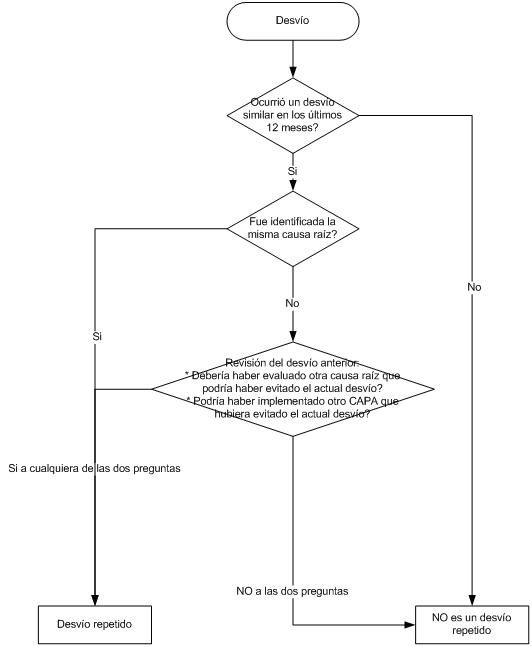
Cualquier consulta sobre el tema, pueden hacerla vía el blog o escribiendo a : info@cgmpdoc.com con el asunto: Desvíos repetidos.
Espero que les sea útil.
¿Cómo conducir una revisión anual de productos efectiva?
Antes de comenzar quiero volver sobre una idea mencionada anteriormente y que supongo la habrán escuchado muchas veces y en distintos lugares: el APR es más que un requerimiento regulatorio, es una revisión que le permite al elaborador entender más su proceso y tomar acciones de mejora.
Las cGMP necesitan una evaluación anual de los estándares de calidad de los productos para determinar la necesidad de ajustar sus especificaciones y los procedimientos de elaboración y de control.
Las guidelines acentúan la importancia del análisis de las investigaciones o desvíos conducidos y los reclamos de productos recibidos, mientras el reporte de APR debe explorar, en profundidad, las causas de recalls y de devoluciones de productos, si las hubiera.
De manera general, el mandato de las Agencias regulatorias (por ej. FDA) es mirar a través y sistemáticamente por todas las áreas de mejora y alinear los procesos consistentemente a la elaboración de productos de calidad.
Por estas mismas razones generales, las GMP requieren a los elaboradores analizar previa revisión, examinar los resultados del análisis de productos terminados y los controles en procesos críticos y revisar: lotes fallidos, desvíos, CAPAs, controles de cambio, estudios de estabilidad, devoluciones, reclamos, calificaciones en equipos críticos y acuerdos de calidad.
Los CAPAs de la revisión anual de productos necesitan ser comunicados al Senior Management y completados de manera efectiva y a tiempo, su efectividad debe ser chequeada por medio de las auditorías internas.
Estructura de un informe de APR:
Puede variar según la compañía, o incluso el producto. La idea de seguir un template o modelo asegura que todos los aspectos requeridos son evaluados.
Pero un APR es un documento que evoluciona.
Estructura de informe de APR: http://wp.me/p1Hn5Y-fs
Cambios y correcciones
La Revisión de los cambios puede ser desglosada en: cambios de materia prima, en los componentes de empaque, en especificaciones y documentos maestros. La sección de no conformidades y desvíos necesita ser revisada sobretodo considerando las acciones correctivas y su efectividad. Cualquier CAPA vencido o inefectivo necesita ser discutido en el informe. Este es uno de los aspectos fundamentales.
Cualquier tendencia observada necesita ser direccionada, no solo las que son OOS.
Racionalización de los datos de origen y administración del APR
Compilar los datos crudos es siempre un esfuerzo de equipo, pero el departamento de QA debe tomar el liderazgo y ser el último responsable de la administración del programa.
Un comité de APR podría típicamente incluir un representante de QA, QC, validaciones, operaciones, estabilidad, ingeniería, y logística. Un borrador de informe es completado sobre el análisis crítico de los datos crudos, luego se discute en una reunión del comité de APR para determinar los CAPAs efectivos.
Otro desafío para el administrador de APR es la recuperación de datos para el propósito de la revisión.
Las empresas con sistemas de adquisición de datos calificados pueden usar sus bases de datos, mientras los elaboradores que se manejan con papeles pueden tener que revisar los documentos de lote en forma individual para ver los parámetros del proceso, los controles en proceso, los análisis finales, los rendimientos, etc. En cualquiera de las dos formas, los datos crudos usados para el análisis deben ser exactos de manera de completar una análisis/evaluación efectivo. Si son observadas desviaciones del proceso durante la revisión, puede ser requerido colectar información adicional para justificar dichos hallazgos.
Los datos deben estar disponibles para el administrador del APR en el momento oportuno. Todos ellos deben ser verificados por una segunda persona si son colectados manualmente.
Si son utilizadas planillas de cálculo, las mismas deben estar validadas previo a su uso.
Conclusiones
Efectuar un APR es un requerimiento para los mercados regulados. Pero más que eso, la revisión ayuda al elaborador a entender y conocer mejor sus procesos y para reunir información adicional para posteriores mejoras.
Es una enorme ayuda en la determinación si un producto aún cumple las especificaciones, si necesita un cambio de formulación, modificación del empaque, una revisión de especificación, o un proceso más robusto. Una conclusión de APR es un paso previo al desarrollo futuro del producto y por lo tanto debería ser exacto y respaldado mediante datos adecuados.
Las guías de validación de procesos de la FDA piden la verificación continua del proceso. Así, un programa de APR puede servir como un sistema on going (etapa 3: verificación continua del proceso) para colectar y analizar los datos del producto / proceso que relaciona la calidad del producto.
Las necesidades del APR son parte del plan de mitigación de riesgos de acuerdo a las recomendaciones del ICH Q9.
La información reunida y las tendencias observadas pueden ayudar al desarrollo de un nuevo producto como tal y entonces esto es esencial para distribuir el reporte a todas las partes pertinentes e interesadas. El esfuerzo puede además ser revisado y compartido con equipos de mejora continua (lean process) mientras el desarrollo de CAPAs de un APR son críticos en evitar riesgos potenciales para un producto en el futuro.
Les dejo un artículo adicional:http://wp.me/p1Hn5Y-f3
Desde cGMPdoc, le ofrecemos la posibilidad de una promoción compuesta de 1 SOP para el manejo de los APR el cual incluye un informe modelo y una capacitación powerpoint con notas aclaratorias y su respectivo cuestionario de evaluación, consulte en info@cgmpdoc.com.
Además si Ud. Lo necesita podemos ofrecerle efectuar la revisión anual de sus productos, consultenos aquí.
La agencia regulatoria Inglesa (MHRA= Medicines and Healthcare products Regulatory Agency). Ha publicado en su página un reporte donde analiza los hallazgos principales sobre un número de 303 inspecciones realizadas durante el último año.
De allí surgieron:
26 observaciones críticas
644 observaciones mayores
Luego del análisis efectuado por la Agencia, quedó consolidado el siguiente listado conteniendo el top 10 de categorías con deficiencia:
1. Investigación de anomalías
2. Quality Management – Control de cambios
3. Investigación de anomalías – CAPA
4. Reclamos y Recall de productos
5. Quality Management
6. Auditoría a proveedores y contratados
7. Contaminación, fisicoquímica (potencial)
8. Documentación – PSF/Procedimientos/Acuerdos Técnicos
9. Documentación – Manufactura
10. Validación de procesos
Les envío el enlace a la presentación, donde además podrán encontrar el desarrollo de cada una de las deficiencias con ejemplos de las situaciones hallados, muy interesante para que podamos ver como estamos posicionados en esos temas.
Estas son unas de las principales causas de observaciones regulatorias reportadas por las agencias más importantes:
Manejo de resultados OOS
Durante el año 2011, 10 empresas recibieron Warning Letters de la FDA debido al manejo incorrecto de los resultados OOS (fuera de especificaciones).
Algunos ejemplos mencionados en el artículo que adjuntamos son:
- “Falla al investigar y documentar resultados OOS…”
- “… Falla para ampliar la investigación a otros lotes del mismo producto farmacéutico que puede haber sido asociada con el error específico o discrepancia.”Les dejo el Link al artículo:
http://www.gmp-compliance.org/eca_news_3142_7413@@menuopt@@.html
y un par de links que pueden serle útiles:
Manejo de resultados OOS: http://wp.me/p1Hn5Y-1n
Contaminación microbiana
La MHRA (Medicines and Healthcare products Regulatory Agency) reportó 31 observaciones de contaminación microbiana el año pasado (2011), frente a las 15 reportadas en el año 2010.
Este aumento lleva a la contaminación microbiana al Top 3 de los defectos de producción más comunes hallados por la MHRA.
Delante de la contaminación microbiana en cuanto a la cantidad de observaciones están:
· La contaminación química o física (algunos ejemplos relacionados con el sistema de HVAC)
· La documentación de fabricación
Les dejo el enlace del artículo:
Investigaciones
Desde 2007, la misma deficiencia, ha encabezado la tabla. Fallas vinculadas a las investigaciones de las anomalías fueron la deficiencia más común una vez más.
Numerosos ejemplos de estas deficiencias se dan, entre ellos:
“Un gran número de investigaciones se observa que no se cerraron de manera oportuna, o donde permanecen abiertos un número de meses más allá del tiempo de cierre estipulado espera”.
Les dejo algunos links que pueden serle de utilidad:
Sistema de manejo de desvío: http://wp.me/p1Hn5Y-3Z
Resolución de problemas: http://wp.me/p1Hn5Y-4T