Estas líneas son una guía para efectuar la calificación de las instalaciones, equipos y servicios existentes.
Existen guías que aportan mucho detalle para la calificación de los equipos nuevos, sin embargo debemos reconocer que cuando nos referimos a la calificación del equipamiento existente, la información es limitada.
En este artículo queremos dejarles reflejado nuestro pensamiento sobre este tema.
No es inusual encontrar que una planta tiene una mezcla de equipos nuevos calificados completamente (DQ, IQ, OQ, PQ) y equipamiento parcialmente o NO calificado.
A través de muchos años de uso, las instalaciones, servicios, sistemas y equipos han mostrado su aptitud para funcionar de acuerdo a distintos requerimientos.
Sin embargo, las cGMPs necesitan que los ítems que tienen impacto sobre la calidad del producto, proceso deben estar calificados. La calificación formal es la base de las actividades relacionadas tales como la validación asociada con la introducción de nuevos productos o la validación de los productos existentes que forma el punto de partida para futuros controles de cambio.
La calificación debería proveer verificación documentada que los parámetros definidos como críticos para la operación y mantenimiento son adecuadamente controlados.
Es esencial que la calificación sea práctica y alcanzable, agregue valor al proyecto y esté concentrada en los elementos críticos del equipo. Es recomendable que un enfoque de Análisis de Riesgos sea usado para definir la profundidad de la calificación.
Siempre debería haber una proporción razonable entre el riesgo para la calidad del producto, la cantidad de mediciones a ser efectuadas y la documentación a ser preparada. Por ejemplo la etapa de manufactura, estado regulatorio y el propósito de uso.
Si miramos los requerimientos regulatorios, nos encontramos que la ICH Q7a indica que las Instalaciones, servicios, equipos y sistemas deben ser adecuadamente calificados para asegurar la integridad de los datos y del producto.
Un lineamiento adicional esta dado por las PIC/S (Pharmaceutical Inspection Co-operation Scheme), donde las mismas indican que aunque no sea posible realizar los detalles de una calificación de la instalación para un equipo establecido ni el enfoque detallado para una calificación de la operación, debería haber datos disponibles que verifique y soporten los parámetros operativos y los límites para las variables críticas de la operación del equipamiento.
Adicionalmente, la calibración, limpieza y mantenimiento preventivo, deberían estar documentados y por supuesto deben existir SOPs de operación y procedimientos de entrenamiento de los operadores para el uso del equipamiento.
Ahora vamos a ver el análisis de riesgo (AR) aplicado a las actividades de calificación.
El AR es un enfoque formal y sistemático para identificar riesgos GMP relacionados al equipamiento y sistemas de soporte. Es una herramienta muy útil que puede ser aplicada a la planta, equipamiento y sistemas los cuales han estado en uso por muchos años.
Es recomendado que las instrucciones de manufactura sean usadas en combinación con los requerimientos generales GMP (ej. Diseño, documentación de riesgos de contaminación, utilización en el producto final) como base para el AR.
Cada etapa de la manufactura puede ser evaluada individualmente con respecto a operaciones / actividades críticas (por ej. Velocidad de agitación, rangos de temperatura, presión, humedad, manejo de producto expuesto, etc.).
Descripciones de procesos, reportes de desarrollo y revisiones de calidad del producto pueden asistir en la identificación de las operaciones parámetros críticos de calidad.
El AR puede ser llevado a cabo usando distintos enfoques, ej. HACCP, FMEA, árbol de decisiones, etc. el AR del equipamiento puede ser efectuado para el tren de equipos o por unidad de proceso individual (ej. Reactor, centrífuga, etc.) y sus sistemas de soporte.
Las unidades de operación podrían ser evaluadas de acuerdo al siguiente esquema:
- La operación impacta directamente sobre la calidad del producto?
- La operación crea datos electrónicos los cuales son la base para actividad GMP relacionada?
- Un malfuncionamiento podría impactar directamente sobre la calidad del producto?
- Tiene instrumentos que miden o controlan etapas críticas del proceso?
- La operación / instalación causa riesgo de contaminación al producto o al ambiente de la planta?
- Los materiales de construcción están en contacto directo con el producto?
Si al final de las preguntas anteriores nos encontramos con algún “Si” entonces la operación es considerada como GMP relevante.
Durante el AR, debemos considerar la probabilidad de ocurrencia y la detectabilidad y las medidas para reducir el riesgo deben ser identificadas.
El AR puede ser efectuado usando un template, donde las etapas de trabajo típicas o actividades para la unidad del proceso son listadas (incluyendo instrumentos y sistemas de control).
Adicionalmente los materiales de construcción en contacto directo con el producto son evaluados y los dispositivos de medición y control críticos son identificados para su calibración.
Ahora veamos las actividades de calificación, en el siguiente flujograma:
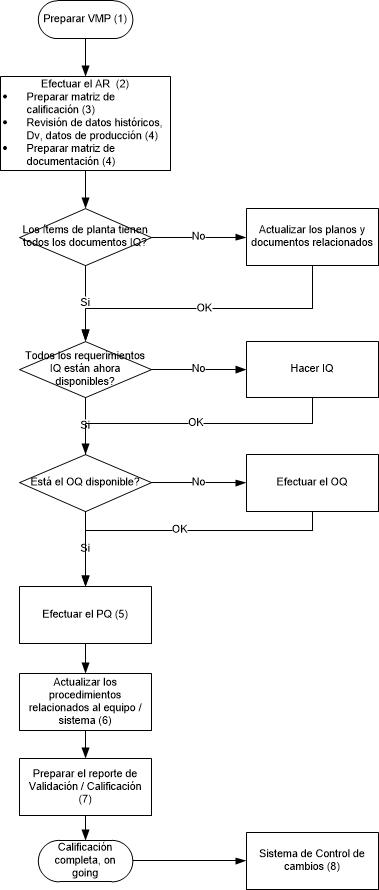
Adicionalmente, los equipos que son calificados, dependiendo de la criticidad de los mismos pueden ser ingresados en un plan de revisión periódica.