El objetivo de este artículo es describir un enfoque científico basado en el riesgo para el manejo de la contaminación cruzada, identificar el nivel de riesgo de contaminación cruzada del producto y determinar las acciones de mitigación apropiadas para la protección de:
- La calidad del producto
- El personal
Para:
- La construcción de nuevas instalaciones
- La modificación de las existentes
- La tercerización de la elaboración de productos
Esto es alcanzado en la práctica por medio de la evaluación y documentación de los riesgos de contaminación cruzada y la toma de acciones de mitigación adecuadas, a lo largo del ciclo de vida del producto entero (La elaboración del producto terminado y su packaging), de manera de asegurar que los productos son adecuadamente protegidos de la contaminación cruzada.
Hay aspectos GMP que deben ser cumplidos, y además hay aspectos relacionados a la higiene industrial y el control de la exposición del personal de manufactura que deben ser considerados.
El aumento del portfolio de productos requiere el manejo de un amplio rango de sustancias, algunas de las cuales pueden ser altamente peligrosas. Esto ha resaltado la necesidad de hacer foco sobre identificar y entender claramente los peligros y cómo se manejarán los riesgos de contaminación cruzada y el control adecuado del potencial para el paciente y la exposición del empleado.
Como mencionamos antes, algunos de los factores que afectan la elección de una planta (nueva o existente) son:
- Portfolio de productos (peligro y riesgo)
- Procesos requeridos
- Restricciones de las instalaciones
- Capacidades de los equipos (contención, facilidad de limpieza y mantenimiento)
Vamos a intentar describir las actividades de evaluación necesarias para evaluar el caso donde múltiples productos puedan ser manejados en las mismas instalaciones. Esto también aplica a la introducción de nuevos productos o a la transferencia de productos establecidos a otras plantas.
Por supuesto que el diseño y estrategia preferido es el flexible, disponer de instalaciones multiproductos, lo cual debe tomar en cuenta tanto los requerimientos GMP/ regulatorios como los de higiene industrial.
Para la gestión de riesgo seguimos los lineamientos de la guía de ICH Q9.
Un enfoque claro es necesario para evaluar los riesgos tanto en las áreas de GMP como de HI y definir sobre un criterio científico de manera de permitir tomar la decisión apropiada para el manejo del riesgo.
Debemos balancear las necesidades GMP y las de Higiene industrial (HI), ya que es muy importante asegurar que todos los riesgos, para el producto y para el operador son adecuadamente manejados.
Es sumamente importante relacionar los conceptos que vemos de contaminación cruzada con un proyecto de inversión y el Gantt de las actividades de calificación / validación, sobre todo cuando pensamos en que las plantas tienen que cumplir las exigencias de las cGMP.
Una primera evaluación de alto nivel puede ser efectuada de manera temprana en el proyecto en la fase justificación, cuando la información del producto y del proceso es reunida. Luego en la fase de diseño, el rango del producto, los límites de limpieza, diseño, layout de instalaciones y selección de equipos necesitan ser considerados desde el punto de vista de contaminación cruzada. Más adelante en el proyecto, debe ser efectuado con más detalle un análisis de riesgo de calidad y el enfoque final de diseño y control debe ser establecido antes de la validación. La verificación de que los riesgos de contaminación cruzada son aceptables, serán alcanzados en la validación de limpieza y otros ensayos de control introducidos (para mix up o transferencia mecánica o aérea).
De manera paralela al análisis de riesgo de calidad (GMP), SHE debe efectuar un análisis de higiene industrial (HI) documentado.
Es recomendable efectuar una comparación y revisión de los dos diferentes análisis de riesgos (GMP y HI) una vez que son completados los mismos, para asegurar que cualquier conflicto sea evaluado, por ejemplo: podría haber un conflicto GMP / HI en relación a la dirección del flujo de aire, típicamente el flujo de aire podría ser bajo presión positiva desde un área de manufactura para propósitos GMP de manera de prevenir la entrada al área, pero una presión negativa para propósito de HI para mantener el producto contenido. El diseño final y la solución de control deberán ser acordadas sobre una base caso a caso balanceando ambos requerimientos (GMP y HI).
Ambos criterios GMP e HI son extraídos desde datos toxicológicos y clínicos de fuentes comunes. Cada uno debe tener un enfoque basado en la ciencia para la gestión de riesgo y una clara estrategia para evaluar la exposición potencial.
A pesar que los criterios para ambos requerimientos provienen de los mismos datos toxicológicos y/o clínicos, la forma en que los datos son usados para obtener límites aceptables y las estrategias de control adecuados difieren.
Resumen de diferencias para consideraciones GMP y de HI
| cGMP | Higiene Industrial (HI) | |
| ¿Quién o qué está expuesto? | Producto | Trabajador (operador) |
| Ruta de ingreso | Contaminación cruzada de producto (por medio polvo asentado o producto X retenido en o sobre el producto Y)
Ingestión del paciente, IV (por vía de administración) |
Inhalación
Dérmica A través de membranas mucosas Ingestión |
| Mecanismos de exposición primaria | Retención (inadecuada limpieza)
Mix UP (materiales erróneos) Transferencia mecánica (movimiento de residuos de un objeto a otro) Transferencia aérea (polvo en el aire y contacta con productos o equipos) |
Inhalación (polvo asentado puede ser re-suspendido para ser respirado en otro momento)
Absorción por piel (por contacto o por vía heridas) A través de membranas mucosas (trabajador contaminado toca sus membranas mucosas) Ingestión |
| Bases de estándares para análisis de riesgo | Límites de limpieza
Exposición diaria aceptable (ADE = Acceptable Daily Exposure) |
Límite de exposición ocupacional (OEL= Occupational Exposure Limit) |
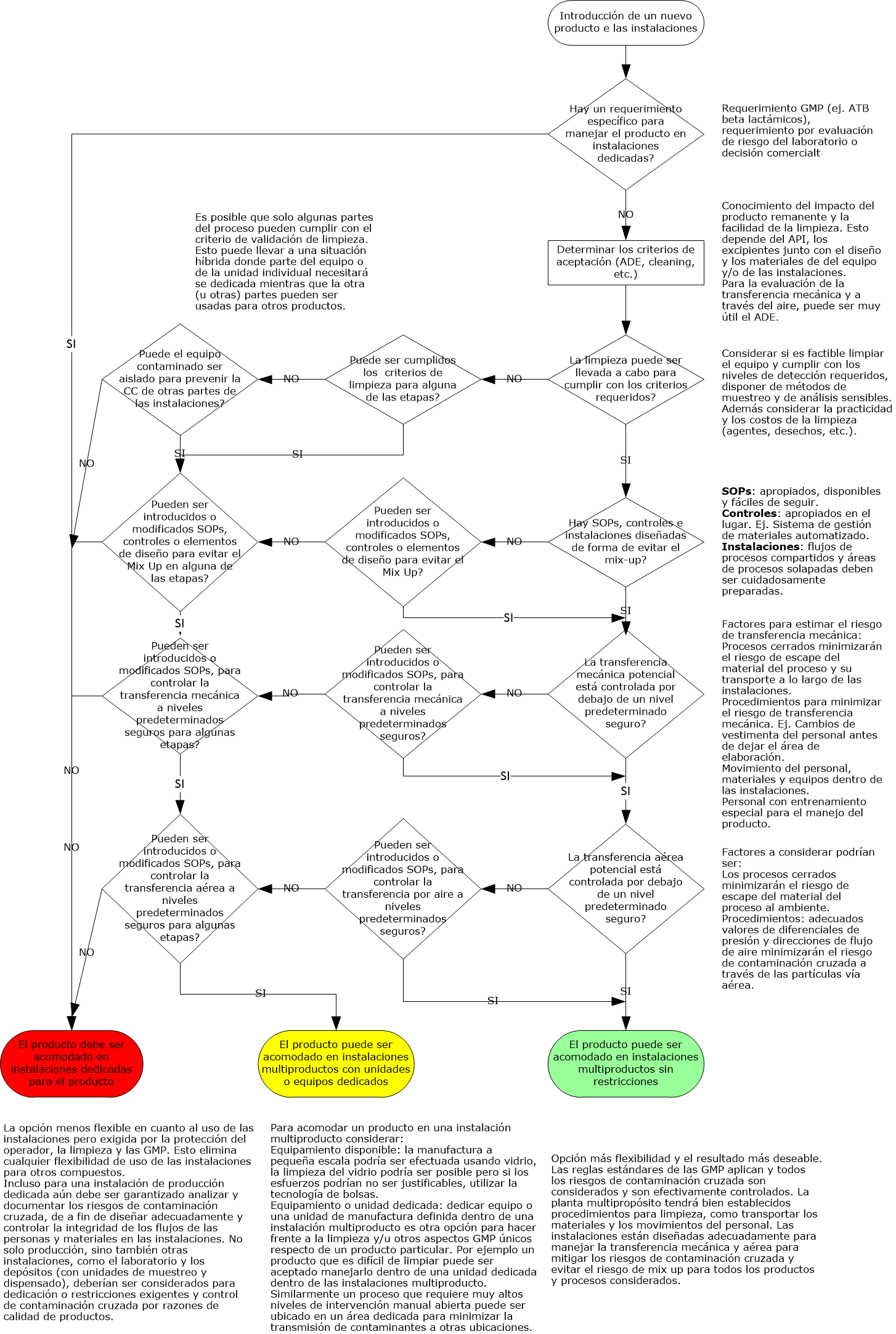
Flujograma GMP de contaminación cruzada GMP: véalo aquí.
Análisis de riesgos GMP
La siguiente información necesitará ser colectada y comprendida para efectuar la evaluación de riesgo:
- Propiedades del producto (ej. Solubilidad, toxicidad, tamaño de partícula) del producto que está en las instalaciones o que será introducido en ellas
- Layout, flujo del proceso, flujo del personal, flujo del aire, diseño de la ventilación
- Diseño del equipamiento y método de operación
- Controles ya existentes en las instalaciones o controles planeados para nuevas instalaciones o productos
- SOPs para control de mix up, limpieza, vestimenta, etc.
Cualquier declaración inicial de alto nivel y decisiones en un proyecto que un cierto producto o tipo de producto no puede ser introducido o empacado en una instalación necesita ser documentado apropiadamente, por ej. En la estrategia de validación y además en las bases del documento de SHE de las instalaciones.
Hay objetivos claves GMP, requerimientos de alto nivel para minimizar el riesgo de contaminación que necesitan ser documentados en URS (requerimientos de usuarios) para asegurar que ellos están incluidos en el proceso de calificación / validación.
Para instalaciones existentes, evaluar que el nuevo producto no será comprometido / contaminado por cualquiera de los productos o procesos existentes en las instalaciones y viceversa, más allá de un límite aceptable.
Para instalaciones nuevas, los productos no serán comprometidos o contaminados en ninguna forma por medio de las actividades de manufactura más allá de un límite aceptable.
Para contaminación cruzada las siguientes cuatro rutas necesitan ser consideradas:
- Mix- Up
- Retención
- Transferencia mecánica
- Aerotransporte
Para ver mas detalle sobre cada una de ellas haga click en Contaminación cruzada.
El impacto sobre el producto / paciente necesita ser evaluado usando la información y conocimientos disponibles.
La competencia en el equipo debería representar especialista con conocimiento en todas estas áreas para que el análisis sea exitoso.
Riesgo: es la combinación de la severidad de las consecuencias potenciales derivada de un peligro (o combinación de peligros) y la probabilidad que esas consecuencias sean realizadas.
Hay diferentes herramientas de análisis de riesgo que pueden ser usadas pero todas ellas deberían incluir factores definidos para evaluar severidad, probabilidad de ocurrencia y detectabilidad y manejar escalas de score comunes.
Severidad: una medida de las posibles consecuencias de un peligro, por ej. Cuán severo podría ser si un paciente / empleado es expuesto a esa peligrosa contaminación? (la escala podría ir desde inadvertido a fatalidad).
Ocurrencia: una medida de la probabilidad que un peligro ocurra. Por ej. Cuán probable es que esa contaminación peligrosa ocurra? (escala desde una ocurrencia en más de 5 años a más de una por lote).
Detectabilidad: la habilidad para descubrir o determinar la existencia, presencia, o hecho de un peligro, por ej. Cuán probable es detectar la contaminación peligrosa antes que la misma sea expuesta al paciente / empleado? (la escala puede ir desde obvio o monitoreado y con alarmas a no detectable).
El propósito del análisis es describir el riesgo, entender la probabilidad de su ocurrencia, si será detectado y evaluar su severidad.
Mitigaciones del riesgo pueden entonces ser determinadas e implementadas. El análisis asistirá en concluir en qué tipo de instalaciones el producto / productos puede ser acomodado. El análisis de riesgo debería ser revisado por medio del proyecto sobre una base regular de manera de asegurar que las mitigaciones son implementadas. El proyecto debería chequear que todas las mitigaciones están “in place” antes de sea entregado para la operación de rutina.
Cuando hay cambios al alcance original del análisis de riesgo el negocio debería revisar el análisis. Si no hay cambios en las instalaciones es recomendado revisar el análisis sobre una base regular, la frecuencia podría tomar en cuenta el portfolio de productos existente.
Espero que le resulte interesante.