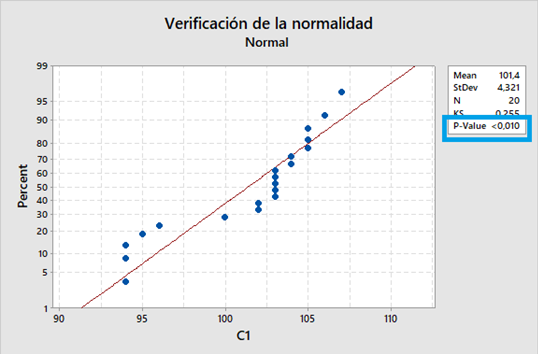
Leyendo un artículo del Pharmaceutical on line, de Peter H. Calcott, D.Phil, tome unos párrafos de la misma que me parecieron sumamente importantes, por la claridad de conceptos sobre algunos temas que usualmente a todos nos generan dudas.
¿Qué escala debo usar en la Gestión de Riesgos de Calidad?
La gestión de riesgos de calidad (GRC) consiste en convertir opiniones en datos o números. Pero lidiar con esos números puede ser desalentador. ¿Qué escala debemos usar? ¿La escala de tres, cuatro, cinco o diez puntos? Tiendo a favorecer los tres o cinco, dependiendo de la situación.
Para un análisis preliminar de peligros (PHA), tiendo a favorecer una escala de tres puntos. Básicamente, es alto, medio o bajo. Es simple y fácil para tomar decisiones. En realidad, trata de no diferenciar la gravedad y la probabilidad entre sí. Simplemente puntúa el riesgo. Esto es útil para un análisis inicial o menor.
Para un análisis más detallado usando FMEA, prefiero una escala de cinco puntos. Da suficientes posibilidades sin ser demasiado complejo. A menudo, con una escala de 10 puntos, los miembros discuten entre un 7 y un 8. Eso simplemente no es importante. Incluso en la escala del 1 al 5, defina lo que cada uno representa para la gravedad y la probabilidad. No es necesario que sea demasiado elaborado, solo una descripción verbal clara. En la mayoría de los análisis FMEA, examinamos la gravedad (¿qué es lo peor que podría pasar?) y la probabilidad (¿qué probabilidad hay de que suceda?).
Estas son mis preferencias porque son simples. Puede usar una escala diferente, y eso también está bien.
La guía ICH Q9 también exige examinar la detectabilidad e incorporarla en los cálculos. Prefiero mantenerlo separado. Hago una evaluación, pero hago el análisis sin ella. Hago mi remediación basado en la severidad y la probabilidad. Si no puedo reducir el peligro lo suficiente, ahí es cuando entra en juego la detectabilidad. Luego, trabajo para que el problema sea detectable. Si no puedo resolverlo, al menos sé cuándo está ahí.
Recuerde, sus números son subjetivos
A medida que realiza los cálculos y la asignación de riesgos, recuerde que los números que asigna son subjetivos. Es la interpretación de sus opiniones, a menudo con pocos datos duros, pero a veces, tal vez solo datos blandos. Trátelos como tales y no se obsesione con los números reales. Estos números no deben tratarse como especificaciones, con asignación de suspenso y aprobación. El propósito de los números es realmente agrupar los elementos en tres cubos. Los primeros son los que deben ser subsanados. El segundo se puede dejar solo. Es decir, no se hace nada. El tercer cubo es para los elementos que se encuentran en el medio. Entonces, hay que preguntarse, ¿Lo arreglo? ¿Lo dejo? ¿O lo observo?
Mucha gente me pregunta: “¿Cómo estableces el estándar para decidir en cuál trabajarás y en cuál no?” Mi respuesta es mirar el análisis y tendrá una idea de dónde, en función de algunos de los elementos que ya sabe intuitivamente que necesitan reparación o no. La mayoría cae en los dos cubos anteriores y la decisión es clara. Es el tercer cubo con el que es más difícil lidiar. Después de hacer el análisis, revise los resultados. Haz la pregunta, ¿se siente bien? Está perfectamente bien decidir remediar algo que puntúa bajo en riesgo o descartar algo que puntúa alto. La clave es documentar sus decisiones, junto con las suposiciones y los datos que utilizó. Este informe se revisará más adelante y es posible que decida cambiar de opinión. La clave es documentar las razones para el futuro.
Al final del día, mire el resultado y pregúntese si se ve bien y tiene sentido. Si la respuesta es sí, ya está. Si no, llévelo a través de otro ciclo.
Elementos importantes a tener en cuenta
Entonces, ha completado el análisis. Eso significa que tiene las herramientas para ayudarlo a priorizar el trabajo y asignar los recursos adecuados para los problemas y las soluciones. Después de la remediación, es aconsejable volver a realizar el ejercicio para asegurarse de que las soluciones han reducido el peligro a un nivel de riesgo aceptable. Incluso entonces, no ha terminado.
Hay otras dos cosas con las que algunas personas tienen problemas. La primera es aceptar que el riesgo siempre estará ahí. Nunca llegarás al riesgo cero. O te volverás loco para llegar allí o te arruinarás. Tienes que aceptar que siempre habrá algún riesgo residual. Lo aceptamos en la vida real. ¿Por qué no aquí?
El segundo es la creencia de que los reguladores no aceptarán su decisión de no hacer nada. De hecho, este miedo se cuantificó en una encuesta que un colega y yo realizamos hace varios años en toda la industria. Un porcentaje considerable de encuestados no estaba convencido de que los representantes de las agencias aceptaran sus decisiones. Pero las agencias han dicho que utilizarán las técnicas. He desarrollado dispositivos, que incluyeron mucho análisis de riesgo en el desarrollo, y he visto a los reguladores aceptar mis análisis de riesgo durante las inspecciones.
Otro elemento, no relacionado con los puntos anteriores, estaba relacionado con una observación que se hizo en una empresa que permanecerá en el anonimato, por razones obvias. Esa empresa le presentó a un colega mío un análisis de riesgos que estaba muy bien hecho y bien documentado. Detalló un análisis de si liberarían un producto basado en un análisis de riesgo. ¡Hasta aquí todo bien! Pero el análisis de riesgo no estaba relacionado con el cumplimiento, la seguridad del paciente o la calidad del producto. Más bien, estaba relacionado con si serían atrapados durante una inspección u otra interacción regulatoria y las consecuencias. Mi colega rápidamente lo cortó de raíz, como dicen. El mensaje para llevar a casa aquí es que el análisis de riesgos nunca debe usarse para justificar algo que usted sabe intrínsecamente que no es correcto.
Espero que les resulte útil, a mi me pareció muy interesante el análisis de Peter H. Calcott, D.Phil.