Hay con frecuencia muchas discusiones en la industria farmacéutica respecto de la naturaleza exacta del proceso de validación. La validación de proceso es un requerimiento de las cGMP y por lo tanto aplica a la elaboración de productos medicinales y dispositivos médicos.
La FDA define validación de procesos como:“establecer evidencia documentada, la cual provee un alto grado de seguridad que un proceso específico producirá consistentemente un producto que cumpla con especificaciones predeterminadas y características de calidad”.
La intención original de la validación fue clara y simple, evidencia documentada que un proceso hace lo que pretende hacer y es exitoso como proceso. Actualmente Plan Maestro de Validación formal es requerido por la mayoría de empresas y este documento contiene enormes detalles, muchos de los cuales son duplicados en los documentos de IQ, OQ y PQ, y los costos de validación en la mayoría de los proyectos superan el 10 % del costo total del proyecto.
La verdadera intención de la validación, tal como está definido en la guía de la FDA, puede haber sido perdida en la interpretación que la industria que tiene que testear 3 lotes con resultados aceptables. Estos tres lotes son probablemente los que tienen menos variación respecto de cualquier de los lotes producidos en la planta, debido a la necesidad crítica de completar la validación de manera exitosa, y los cuidados de la manufactura en esos tres lotes de validación.
Sin embargo, para obtener el conocimiento necesario de los productos y procesos, y al mismo tiempo producir un retorno de la inversión más atractiva, la validación debe basarse en la ciencia.
Si un modelo DOE se utiliza tempranamente en el proceso para caracterizar el producto y el proceso, el modelo confirmado puede luego ser usado para establecer límites óptimos para maximizar el resultado y minimizar la variación y predecir que podría suceder bajo las condiciones alternativas.
Posts tagged ‘cGMP’
Durante bastante tiempo hemos tenido en mente que los procesos de validación y de calificación debían contemplar dentro de los procedimientos cuál era la frecuencia con que debíamos repetir la actividad, sin embargo las cGMP nos van orientando hacia implementar una serie de actividades sistemáticas que nos permitan revisar y documentar que nuestros equipos, procesos, sistemas, etc. mantienen el estado validado o calificado. Por ejemplo en sistemas computarizados hablamos de efectuar una revisión periódica de los sistemas computarizados validados, en el caso de los procesos validados hablamos de seguirlos a través de la verificación continua del proceso o de la Revisión Anual de Productos.
¿Cuáles son los ciclos adecuados para la recalificación de equipos?
Con la revisión del Anexo 15 en octubre de 2015, el tema de la recalificación se ha vuelto más importante. En el antiguo Anexo 15 de 2001, el tema de la recalificación estaba “oculto” bajo la demanda general de revalidación (punto 45). Con la revisión del Anexo 15, la recalificación ahora tiene su propio capítulo con los requisitos para:
- Una evaluación del equipo con la frecuencia adecuada para demostrar que se ha mantenido en un estado de control y
- Cuando es requerida una recalificación, los intervalos de tiempo deben justificarse y los criterios para la evaluación deben establecerse.
En la práctica, a veces es difícil establecer tales programas y criterios para la evaluación. En el área estéril, a veces hay referencias específicas de las regulaciones a dispositivos y procesos individuales. Por ejemplo, de acuerdo con la Guía aséptica de la FDA, los filtros HEPA en sala limpia clase 5 deben probarse dos veces al año. El Anexo 1 de las Directrices GMP de la UE también proporciona directrices sobre este tema (por ejemplo, para la esterilización).
¿Qué pasa con el equipo en otras áreas?
En este caso, puede ser útil el capítulo 9 de la línea de base ISPE n° 5 revisada Puesta en servicio y calificación de junio de 2019 sobre la revisión periódica. El enfoque de “revisión”, es decir, la evaluación, se presenta en dos fases.
En la fase 1, se clasifican los “sistemas de impacto directo” existentes. Dependiendo de la complejidad del sistema (complejo vs. estándar) y la influencia en la calidad del producto, los sistemas se dividen de manera ejemplar en las categorías 0-3. Cada categoría, excepto la categoría 0, se asigna a un período de “revisión”. La categoría 0 se refiere a los sistemas existentes donde tenemos disponibles datos de monitoreo. En este caso, no es necesaria una revisión de estos sistemas de categoría 0 (por ejemplo, sistemas de agua). Ya que tenemos datos para su evaluación. Para sistemas en la categoría 1, por ej. autoclaves, se aplican las especificaciones anteriores de las reglamentaciones estériles. Para sistemas de categoría 2, por ej. tanques de almacenamiento intermedio, la línea de base sugiere un intervalo de dos años. Para sistemas de categoría 3, por ej. comprimidoras, la línea de base recomienda un intervalo de revisión de tres años.
En la fase 2, se ejecuta la “revisión”. La revisión en sí tiene lugar como un proceso de tres pasos:
Paso 1: una evaluación inicial con respecto al cumplimiento de GMP, historial de cambios, mantenimiento / calibración y desviaciones.
Si esta evaluación plantea dudas sobre el impacto del sistema en la calidad del producto, se debe seguir el paso 2. De lo contrario, el proceso termina aquí.
Paso 2: en función del resultado del paso 1, los expertos en la materia (SME) examinan en mayor profundidad los sistemas examinados en el paso 1, cuyos resultados son cuestionables. Los SMEs deben provenir de los mismos departamentos que también participaron en la evaluación en el paso 1.
Si la evaluación en el paso 2 llega a la conclusión de que un sistema ya no puede estar en estado calificado, el siguiente paso, el paso 3, tiene lugar. De lo contrario, el proceso termina aquí.
Paso 3: en función del resultado del paso 2, los SMEs examinan en mayor profundidad los sistemas examinados en el paso 2, cuyos resultados son cuestionables. Los SMEs deben provenir de los mismos departamentos que también participaron en la evaluación en los pasos 1 y 2. Si es necesario, se deben tomar medidas para que el sistema vuelva a un estado calificado. Se debe crear un documento de desviación para controlar las acciones tomadas.
De acuerdo con la línea de base, todo el proceso debe ser administrado por alguien con una gran experiencia. La revisión en sí misma debe al menos ser aprobada por el propietario del sistema y la unidad de calidad. Un formulario de revisión es parte de la línea de base como Apéndice 11 y se da un ejemplo en el Apéndice 12.
Tomado de la news Letter de la ECA (28/8/2019)
El objetivo de la Revisión de Batch Record (BRR) no es simplemente identificar excepciones (errores, olvidos, entradas ilegibles, etc.), tener el registro corregido oportunamente provee una documentación exacta de las etapas que comprenden la manufactura o el empaque del lote en cuestión.
El BR puede ser requerido semanas, meses o incluso años después, para buscar información. Además puede ser solicitado por parte de la agencia, por lo tanto necesitan ser corregidos y ser claros antes de ser archivados.
Si bien la importancia de la Revisión de BR es indiscutible, hay desafíos logísticos en el proceso de corrección. Esto surge desde la necesidad de controlar la BRR y la disponibilidad del personal requerido para hacer las correcciones. Idealmente, todos los errores ingresados deben ser identificados en el departamento revisor antes que el BR vuelva a Aseguramiento de Calidad (QA) para la revisión de calidad.
La revisión debe ser para identificar cualquier información faltante y que las entradas sean correctas y están dentro de los parámetros establecidos.

Asumiendo sin embargo, que hay una pregunta o corrección requerida a nivel de la revisión de QA, la persona que hizo la entrada original (o falla al hacer la entrada) sobre el BR, cumple con el revisor que identificó la excepción. Esta pregunta o corrección requerida será asentada en el BR para permitir cualquier persona subsecuente entienda el problema.
Cuando una corrección inmediata nos es posible debido a un tema de turno, persona de vacaciones, licencia, etc. entonces el revisor debe informar al supervisor o jefe de la situación para la resolución de la misma.
Hay ocasiones donde falta información vital, la cual no puede ser corroborada a través de registros electrónicos u otros registros. El revisor entonces tiene la responsabilidad de informar esto a quienes son responsables del desvío y la investigación interna. Esta etapa debe ser anotada o remarcada en el BR (debe ser indicada una referencia a la investigación). La terminación de la revisión del BR se demorará hasta que la investigación sea completada y la disposición de la desviación es determinada. La revisión debería ser completada y firmada aún si el batch será retrabajado (si está permitido) o destruido. El BR y el check list y la planilla de correcciones, debe entonces ser enviada al área de QA y el estatus de revisión actualizado en el log o base de datos.
Revisión periódica de los BR revisados
De acuerdo a nuestra experiencia, es de mucha utilidad efectuar una revisión periódica por medio de la supervisión o jefatura o gerencia, dentro de la unidad de calidad para determinar que se está alcanzando la consistencia deseada en el proceso de revisión. Una revisión de las planillas de corrección provenientes de varios revisores proveerá una perspectiva útil sobre el número de excepciones citadas y / o correcciones requeridas, los tipos de excepciones citadas o las correcciones procuradas, y una comparación de los hallazgos de los distintos revisores. La tendencia de los resultados de producción es una herramienta muy útil para detectar desvíos en el proceso de manufactura y a partir de ellos identificar etapas de corrección para ser tomadas. Si el número de correcciones es elevado, entonces uno debe cuestionarse si el personal de manufactura y los revisores de los BR tienen el mismo conocimiento o entendimiento de que constituye una documentación de BR exacta y suficientemente completa. Si existe un desentendimiento, tal vez el entrenamiento inicial para el personal de manufactura, personal calificado, o ambos no fue lo suficientemente detallado, o el lenguaje del BR es ambiguo y debido a esto, puede ser malinterpretado. El objetivo es asegurar que el personal de manufactura y los revisores están trabajando hacia el mismo objetivo de buena documentación.
Desde una perspectiva ligeramente diferente, si el tipo de excepciones o correcciones requeridas es alto para un BR particular, entonces el lenguaje de dicho MBR debería ser cuidadosamente examinado. Si el lenguaje aparentemente es claro, entonces la documentación en cuestión debería ser revisada con el personal de manufactura. La variación es observada principalmente en un turno? Es requerido un entrenamiento adicional? Si el lenguaje es ambiguo, tal vez la próxima etapa es siguiendo los lineamientos del SOP de control de cambios modificar el BR y luego reentrenar el personal.
Finalmente, hay una discrepancia entre los revisores individuales sobre el número y los tipos de excepciones o correcciones observadas? Esta determinación puede llevar a una discusión muy útil y de fina sintonía para hacer el proceso de revisión más consistente entre los revisores.
A continuación les voy a describir 3 situaciones de inconsistencias en el proceso de revisión, las cuales pueden ser detectadas por medio del proceso de análisis periódico y corregido por medio de la intervención de los supervisores.
- Conveniencia vs exactitud
Si alguna vez han revisado BR, saben que es una tarea tediosa, repetitiva, y demandante. Las etapas requeridas para obtener correcciones de documentos requiere tiempo adicional y seguimiento, un revisor puede caer en el hábito de minimizar el número de correcciones requeridas de un documento como una forma de cerrarlos más rápidamente. Esta es una peligrosa tendencia porque los atajos tienden a convertirse en hábitos, y la inconsistencia introducida por medio de la no revisión de todas las partes del BR puede llevar a dos situaciones indeseables: primero, la exactitud del registro y la información que provee se convierte en sospechosa y segundo, el personal de manufactura se priva de la oportunidad de aprender de los errores o equivocaciones que podría conducir a mejoras.
- Desvío no intencional
Aquí un revisor bien entrenado y experimentado ha revisado muchos BRs que tienen desvíos no intencionales respecto del estándar interno. Mientras el revisor tiene acceso al SOP y al check list para la revisión de los BRs, esos documentos de guía no son para él necesarios, a pesar que el check list es firmado. El proceso de revisión ha tomado una actividad robótica durante la cual el proceso mental no está completamente comprometido, pudiendo haber ausencia de datos o ingresos de datos incorrectos. Esta inconsistencia puede emerger cuando auditorias periódicas de las revisiones conducidas por varios revisores muestran muy diferentes hallazgos.
- Exageración (exceso)
Mientras la falta de plena atención en el proceso de revisión es una fuente de inconsistencia, así también es una inconsistencia el revisor que va mucho más allá del estándar interno. En este escenario, el revisor insiste en que cada detalle, incluso lo que no tiene impacto, sea corregido de acuerdo a su estándar. Esto puede incluso incluir desafíos a la gramática o la ortografía de aquellos que han escrito un comentario satisfactorio o que han corregido en el BR. Algunos revisores pueden indicar algo como: “si Ud. Quiere que yo firma este registro, el mismo debe estar completamente correcto” o “No estoy completamente conforme con este registro”.
Esto no solo consume mas esfuerzos adicionales del personal de manufactura, sino también envía un mensaje desafortunado para aquellos que hacen el producto. La aceptabilidad de un registro aparentemente depende más sobre quien revisa el BR que sobre como el mismo fue completado.
Cualquier inconsistencia en el proceso de revisión de BR desafía la reputación de la unidad de calidad y su gestión, porque demuestra una falta de la vigilancia adecuada de su equipo. La vigilancia es uno de los roles claves de la unidad de calidad en las organizaciones. La gestión de la unidad de calidad asegura que su personal además cumple consistentemente con los estándares establecidos.
Los tipos de controles y los registros y reportes que soportan la manufactura de productos terminados están bien definidos ambos en términos de requerimientos regulatorios y en la práctica de la industria. La revisión adecuada de los BR ejecutados sirve al menos para dos propósitos importantes. El proceso satisface los requerimientos regulatorios y el proceso provee feedback útil para el grupo funcional responsable de manufactura.
Como indicamos, una alta tasa de error puede indicar inadecuado entrenamiento o supervisión pobre, pero además podría indicar un procedimiento escrito pobre. Apropiados revisores y aprobadores de BRs entrenados proveen un valor de servicio de la unidad de QA a la organización de manufactura. Su entrenamiento debe incluir un objetivo de consistencia en el proceso de revisión en adición a la exactitud y minuciosidad, esto puede ser mejor determinado por medio de la revisión periódica de los hallazgos de los revisores.
Espero que les resulte útil.
Dentro de estos requerimientos básicos, les dejamos 10 tips claves, esenciales para asegurar la integridad de los datos dentro de los límites de un sistema de datos electrónicos:

- Identidad única para cada usuario:
Para sistemas basados en papel, siguiendo las Buenas Prácticas de Documentación la identificación de cada analista puede ser alcanzada a través de que cada persona firme o coloque sus iniciales sobre una impresión o notebook de laboratorio. Con los sistemas electrónicos, este principio debe ser mantenido por medio de la identidad única del usuario.
Las cuentas de usuarios no deben ser compartidas, por ej. Una cuenta de usuario establecida para dos analistas para el acceso a un software de laboratorio de un sistema HPLC.
Las identidades de los usuarios no deben ser reusadas, si una persona deja el laboratorio o la compañía, esa identidad del usuario (ID) debe ser removida del uso y cualquier nuevo usuario debe ser habilitado con un nuevo y único número de cuenta de usuario.
- Implementar controles de passwords adecuados:
Los passwords deben ser efectivos de manera que no puedan ser adivinados por otros, por ej. Su nombre / dirección / fecha de cumpleaños, etc. pero no lo demasiado complejos de manera que el usuario lo tiene que tener escrito para poder recordarlo.
Los passwords NO deben ser compartidos bajo ninguna circunstancia. Cuando las credenciales de registro son compartidas, los individuos específicos no pueden ser identificados a través de la actividad de registro y por consiguiente se pierde la integridad de datos.
- Establecer diferentes tipos de usuarios con diferentes privilegios de acceso:
Los accesos a los sistemas o equipos deben ser limitados solo a individuos autorizados, por ej. Analistas, administradores.
Cada tipo de usuario debe ser asignado con diferentes privilegios de acceso de acuerdo al rol que efectuará en el sistema. Debe haber protocolos de seguridad de datos en los cuales se describe el rol del usuario y sus responsabilidades en términos de privilegios de acceso, cambio, modificación, creación y eliminación de proyectos y datos. Solo personal senior debe tener cuentas que le permiten administración completa del sistema, incluyendo la edición de los métodos y de los proyectos.
El rol del administrador del sistema debe asegurar que cualquier requerimiento de cambio, eliminación, etc. es cuidadosamente registrado, y atribuido a un individuo para asegurar que la trazabilidad es mantenida.
- Establecer y mantener una lista de los usuarios actuales e históricos:
Una lista de los usuarios del sistema actuales y de los previos necesita ser establecida y mantenida, si un miembro del personal ha dejado de serlo, entonces la cuenta debe ser inmediatamente puesta como no disponible.
- Control de cambios del sistema:
Relacionado con la asignación de privilegios de accesos está la capacidad de un usuario de hacer cambios. Los cambios deben ser solo efectuados por medio de los individuos autorizados, ej. Cambios de métodos, parámetros de integración y línea de base. Esto debe ser posible para identificar cuáles individuos hicieron los cambios para determinar si ellos tienen el apropiado entrenamiento, educación y experiencia.
- Solamente personal entrenado debe operar el sistema:
Hay un requerimiento para que todo el personal tenga la combinación de educación, entrenamiento y experiencia para efectuar su trabajo. Una parte clave de cualquier programa de entrenamiento es que debe hacer foco sobre la generación de datos y que los cambios solo pueden ser hechos de acuerdo a procedimientos predefinidos para prevenir una acusación de falsificación o fraude.
Los usuarios de instrumentos o equipos deben poder demostrar su capacidad de uso frente a las agencias regulatorias cuando es requerido, dentro de su descripción de rol.
- Registrar toda la información
Los registros de laboratorio deben incluir datos completos derivados de todos los ensayos necesario para asegurar el cumplimiento con las especificaciones y estándares establecidos. Esta es la clave para establecer la integridad de los datos en un sistema de captura de datos electrónicos, a pesar de las situaciones que nos pueden ocurrir, errores, mal funcionamiento de un equipo, etc.
Los datos crudos, “raw data”, (ej. Cromatogramas, pesadas del estándar y de muestras, cálculos, estándares, reactivos, información del instrumento) deben estar disponibles siguiendo la ejecución del trabajo para permitir que una investigación pueda ser llevada a cabo eficientemente y para confirmar la autenticidad y confiabilidad de los datos durante la inspección efectuada por la agencia regulatoria. Todos los datos deben ser registrados de manera que las actividades puedan ser cuidadosamente reconstruidas.
- Definir y documentar registros electrónicos para el sistema
Para registros electrónicos los usuarios deben determinar cuáles datos serán definidos como “raw data”, como mínimo, todos los datos sobre los cuales se basa la toma de decisiones de calidad deben ser definidos como raw data.
Toda la documentación creada durante el análisis la cual incluye un registro es considerada como evidencia de una actividad, de este modo, los archivos de datos creados durante una corrida analítica son registros.
Muchos documentos (instrucciones y/o registros) pueden existir en formatos híbridos, por ej. Algunos elementos como electrónicos y otros en papel. No importa si un registro es generado en papel, existe con firmas manuscritas y le sigue una impresión de un registro electrónico (sistema híbrido) o si es mantenido completamente electrónico.
Controles apropiados deben están en uso para asegurar la integridad de los registros a lo largo del período de retención. Debe estar claramente definido cual registro está relacionado con cada actividad de ensayo y donde es localizado el registro. Controles de seguridad deben estar en uso para asegurar la integridad de los registros a lo largo del período de retención y validado cuando sea apropiado.
- Revisión de las entradas al audit trail para cada lote
Parte de los datos completos de un sistema de datos cromatográfico en las corridas analíticas incluyen el audit trail, y hay un requerimiento bajo las cGMP para la firma o iniciales de una segunda persona para mostrar que el trabajo ha sido hecho correctamente y conforme a los estándares.
Los cambios no deben tapar los datos registrados previamente (sobre escritura).
- Copia de seguridad del Sistema regular
Copias de seguridad (BackUp) regulares de toda la información relevante debe ser llevada a cabo. Típicamente esto se hace diariamente. La integridad y la exactitud de los datos de back up y la capacidad para recuperarlos datos debe ser verificada durante la validación y monitoreada periódicamente. Cuando el back up es efectuado, los registros del back up deben ser chequeados para ver cuando fue efectivo. El back up necesita ser validado, y periódicamente debe efectuarse una recuperación de los datos para ver que el hardware, por ej. CDs, son aún leíbles.
Continuando con el tema de las auditorías, generalmente al desarrollar el tema surgen algunas preguntas, cómo quién o qué debe ser auditado, quién debe ser el auditor, donde y cuando ha de ser efectuada la auditoría y finalmente el porqué de una auditoría GMP, en las próximas líneas vamos a tratar de dar respuestas a las mismas.
- ¿Quién debería ser auditado?
Además de los procesos de Auditorías internas requeridas por las cGMP, los Proveedores (internos y externos) deberían ser auditados para ver su cumplimiento con los requerimientos de las regulaciones GMP y con los estándares de la industria. Dentro de los proveedores podemos mencionar (a modo de ejemplo) a quienes nos suministran:
- Activos (APIs) (estériles y no estériles)
- Productos (estériles y no estériles)
- Excipientes
- Componentes de empaque (en contacto con producto y sin contacto con producto cuando es requerido)
- Materias primas
- Contratistas / elaboradores
- Proveedores de servicios críticos
Además las auditorías nos permiten evaluar un potencial o nuevo proveedor o vendedor, evaluar cambios claves en los proveedores / vendedores o contratistas o investigar problemas específicos que hayan experimentado para determinar la causa.
Estas auditorías determinan el nivel de calidad y cumplimiento de una operación particular.
La auditoría debe ser llevada a cabo siguiente un programa de auditoría.
- ¿Quién es responsable de la auditoría?
El responsable de calidad del laboratorio es quien debe disponer de un plan de auditorías y de personal responsable de ejecutarlas. Dicho plan debe ser seguido en su ejecución, así como los hallazgos de los proveedores a través de un CAPA Plan aprobado por el auditor.
- ¿Cuándo son efectuadas las auditorías?
Hay auditorías que son efectuadas como respuesta a un evento relacionado a compliance.
Generalmente la frecuencia de la auditoría es definida luego de efectuar un análisis de riesgo del proveedor. Variaciones a esta frecuencia definida en el Análisis de Riesgo pueden ser permitidas sobre la base de la performance del proveedor.
Las auditorías de QA deben ser efectuadas sobre una base regular para asegurar que los sistemas de calidad están bajo control y en cumplimiento.
- ¿Por qué efectuar auditorías GMP?
Efectuar auditorías de calidad de productos, procesos y sistemas, es importante para asegurar que todos los laboratorios y los proveedores que dan soporte a las operaciones de los mismos, son revisadas para verificar que están alineados y en cumplimiento con las cGMP.
Una auditoría GMP es una herramienta de evaluación para:
- Verificar si las políticas locales o del laboratorio y los procedimientos están siendo seguidos.
- Verificar que los sistemas y controles requeridos están in place e in compliance con las regulaciones GMP.
Los objetivos específicos de la auditoría GMP son:
- Determinar si las actividades de QA, de producción y demás sistemas cumplen con las GMPs, regulaciones de las agencias regulatorias y los requerimientos de los laboratorios.
- Identificar de forma temprana de problemas de las instalaciones
- Ayudar a determinar la profundidad del problema identificado en el área como también el alcance del mismo en la empresa.
- Investigar y determinar la causa raíz del problema o deficiencia.
- Asegurar compliance con los requerimientos de las agencias regulatorias
- Dar información del nivel del cumplimiento de la planta y de los proveedores respecto de las cGMP.
- Recomendar aprobar y certificar un proveedor.
Cada auditoría que es conducida debe estar basada en hechos.
Los auditores son buscadores de hechos!
Toda observación de una auditoría debe tener disponible la referencia a las GMP y el estándar de la industria actual. Una auditoría debe estar balanceada e incluir las buenas prácticas como también las desviaciones. Si son encontradas desviaciones, las mismas deben ser reportadas. Una auditoría es una herramienta para mejorar las operaciones del laboratorio o las operaciones del proveedor para asegurar que los productos del laboratorio son de un grado de calidad, efectividad y pureza aceptables.
Los beneficios tangibles y a largo plazo de una auditoría incluyen:
- Identificar, eliminar y prevenir problemas de una forma temprana. En el diseño y vida de un proceso o sistema a través de la proactividad.
- Disminuir el número de descartes, recalls, y lotes reprocesados o retrabajados.
- Disminuir el número de reclamos de clientes.
- Mejorar continuamente los sistemas GMP y potencialmente transferir las mejoras prácticas a través de todos los sectores del laboratorio y sus socios.
- Proporcionar una gestión con feedback objetivo basado en hechos que puedan ayudar a identificar debilidades críticas que requieran recursos (capital y/o personal). Efectuar un Risk assessment.
- Asegurar que los clientes del laboratorio reciben productos de una calidad, eficacia y seguridad aceptables.
Recuerde que el auditado es el primer beneficiario de la auditoría.
Ya en otro artículo hemos definido desvío o no conformidad, voy a mencionar otra forma de definirlo:
Un evento no planeado que puede provocar el alejamiento de un proceso, procedimiento, resultado inesperado, no cumplimiento de un requisito previamente establecido en las cGMP.
También mencionamos que los podemos clasificar en críticos, mayores y menores de acuerdo a su impacto o gravedad.
Ahora no voy a volver sobre el proceso de investigación de los desvíos sino que quiero que veamos el tema de la eficacia de las investigaciones.
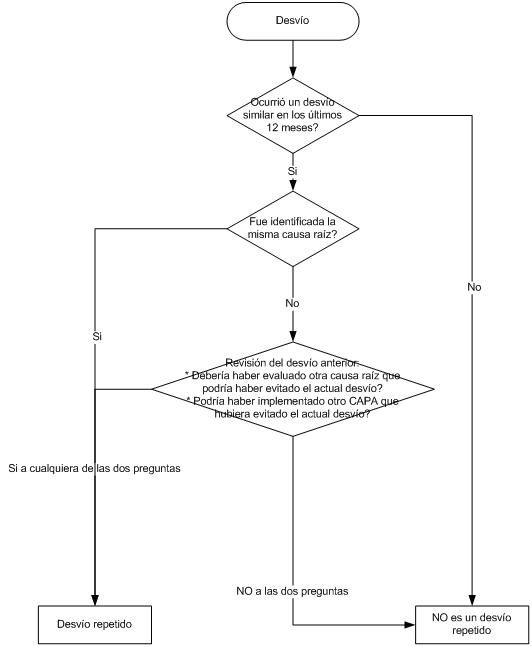
Si bien muchos utilizan la evaluación de la eficacia de los CAPAs, tomándose un tiempo posterior a la implementación del CAPA y revisando los resultados, hoy quiero referirme a la identificación de los desvíos repetidos.
La evaluación del desvío para ver su reincidencia, se hace sobre un período de tiempo (puede ser 12 – 24 meses).
Un desvío repetido es un evento que se repite con la misma causa raíz de cualquier clase de desvío para el mismo o similar producto / material, proceso, equipo, área, documentación u otro sistema GMP dentro del período de tiempo establecido (12-24 meses).
Pero cual es la interpretación de este hecho?:
La ocurrencia de un desvío repetido indicaría que la investigación inicial del desvío no parecería haber sido adecuada en la identificación de la causa raíz del evento o no se completó adecuadamente la acción correctiva para prevenir su reocurrencia.
Para los interesados, les dejo un flujograma muy simple para la identificación de los desvíos repetidos:
Cualquier consulta sobre el tema, pueden hacerla vía el blog o escribiendo a : info@cgmpdoc.com con el asunto: Desvíos repetidos.
Espero que les sea útil.
Rara vez los problemas relacionados al incumplimiento de las cGMP están asociados a empleados deshonestos o proveedores que intentan intencionalmente debilitar los procesos o productos de la compañía. En estos casos las acciones a tomar son fáciles, y pasan por el reemplazo de cualquiera de las dos partes.
Sin embargo con mayor frecuencia, esto se debe a situaciones sistemáticas que pueden ser por conocimiento inadecuado, pobre entrenamiento e insuficiente supervisión.
Una compañía puede mostrar un programa de entrenamiento sólido durante años, sin embargo la capacitación es tan buena como los resultados que produce.
El inadecuado entrenamiento está dentro de los top de incumplimientos de acuerdo a la evaluación de los últimos años efectuada por la FDA.
Ahora muchas veces no preguntamos: ¿Porque no se incorporó el entrenamiento?
Una breve versión diría: Cuando la conducta del trabajador es consistentemente de no cumplimiento, el entrenamiento resulta ser inefectivo.
Algunas preguntas a esta situación de incumplimiento pueden ser:
¿Porque no hizo el trabajo? o ¿No sabían que no estaba funcionando?
Ambas indican un gap peligroso que no puede ser cubierto por el simple agregado de otro curso.
Es importante efectuar un análisis ubicando tendencias, por sector, por personal, por turno, etc. identificando la causa raíz. Y buscando además la comprensión de las personas de la importancia del mismo.
Los programas de capacitación para ser realmente eficaces, deben ser continuamente revisados, repensados y adaptados para satisfacer las necesidades de la industria, su empresa y el estudiante individual.
Les dejo unos links de artículos relacionados al tema:
- Capacitación http://wp.me/p1Hn5Y-2S
- Efectividad del entrenamiento http://wp.me/p1Hn5Y-2X
- Verificación de la tarea por una segunda persona: http://wp.me/p1Hn5Y-3f
- Piense antes de actuar: http://wp.me/p1Hn5Y-4b