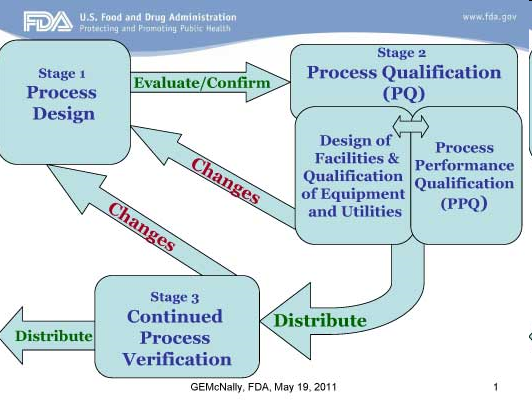
Desde finales de los años 70, “Validación” es una de las palabras GMP más poderosas que pueden usar los reguladores y en los informes de inspectores o auditores. O, mucho más precisamente, las dos palabras “no validado” que provocan un caso de discusión largo y probablemente litigioso entre la industria y las agencias. ¿Por qué esto? Probablemente porque estar involucrado en la protección de la vida y la protección de la salud, nuestra industria no tiene el derecho ético de empeorar la situación de ningún paciente. Luego se considera un producto “validado” (es decir, un producto obtenido a través de un proceso validado) como la calidad mínima que la industria farmacéutica tiene para ofrecer. Las personas que usan productos farmacéuticos deben estar seguras de que se curarán o encontrarán alivio sin ningún efecto adverso derivado de los procesos de fabricación. Aquí, la validación se utiliza para demostrar que nuestra industria ha funcionado bien en la producción de productos efectivos y cualquier falla durante una validación puede realmente considerarse como un problema crítico. El nuevo paradigma de validación sobre la verificación en curso ahora es obligatorio para los nuevos productos y es más que alentador para los productos heredados (tradicionales) en ambos lados del Océano Atlántico por la FDA y la EMA. Sin embargo, dado que no todos los procesos pueden convertirse al nuevo enfoque QbD, la validación tradicional del proceso como se describe en la reciente guía de la EMA debería seguir siendo aceptable durante los próximos años o incluso décadas, y el dossier incluirá: “la descripción del proceso de fabricación, las pruebas que se realizarán y los criterios de aceptación, una descripción de los controles adicionales implementados y los datos que se recopilarán ”. Uno podría decir “como de costumbre” …
Guía de Buenas Prácticas de Validación de la ECA.





{kind=link}