Antes de comenzar con la guía rápida de FMEA, creo que sería bueno revisar el proceso general.
Visión global del Proceso
- Es deseable un buen entendimiento del proceso.
- Los procesos largos, deben ser fraccionados en subprocesos, en etapas o componentes.
- Cada punto o modo de falla de cada etapa o componente, debe ser identificado.
- Luego debe ser identificado el efecto de cada uno de los puntos o modos de falla.
- Finalmente es evaluado cada uno de los simples puntos de falla, para ver su riesgo y la necesidad de acciones de mitigación.
Hay diferentes herramientas para efectuar dicho análisis, nosotros hemos optado por el uso del FMEA (o en español AMFE = Análisis de Modo de Falla y Efectos).
A los interesados en el tema, disponemos de una Planilla Excel, que les servirá para llevar adelante el análisis de riesgo con FMEA, donde en una de las solapas encontrará los criterios de aceptación para cada uno de los 3 criterios utilizados y en la otra una planilla para registrar las evaluaciones efectuadas, solicítela enviando email a: info@cgmpdoc.com.
El FMEA utiliza 3 criterios:
- Severidad
- Probabilidad
- Detectabilidad
Los criterios están establecidos en un rango numérico, los valores altos implicando un mayor impacto. El rango utilizado puede ser bajo, como por ejemplo de 1 a 3 (Bajo, Medio o Alto) para usar la herramienta de forma más simple, sin embargo lo más común es usar un rango de 1 a 5 o de 1 a 10, dependiendo de la definición deseada del score. Un estudio largo con muchos eventos que evaluar debería estar adecuado con largos rangos de valores.
Para cada etapa o componente del proceso, determinar el o los modos de fallas y cómo la falla podría ser presentada por sí misma (efecto de la falla – qué defecto es creado).
La pregunta en esta etapa es ¿Qué podría salir mal?
Luego para casa caso de falla, es importante determinar el impacto, el efecto o el riesgo.
Para cada uno de estos casos, se determinan los 3 criterios anteriores (severidad, probabilidad y detectabilidad).
Severidad
Es una medida de las consecuencias / impacto si el evento ocurre, ¿Cuáles son las consecuencias si esto sale mal?
- Si el riesgo identificado ha sucedido, ¿Cuál es el impacto para el usuario final o para el proceso?
- NO incluir o considerar cualquier mitigación o control actual, o método de detección en el racional cuando se determina el rango de severidad.
- El peor escenario debería ser usado si hay una duda entre dos scores, el score más alto debe ser usado.
Probabilidad (Ocurrencia)
Es una medida de la probabilidad de que el evento suceda. ¿Cuál es la probabilidad que esto salga mal?
- ¿Cuál es la probabilidad que el riesgo ocurra o cuál es la historia de ocurrencia del evento?
- ¿Hay datos históricos que pueden ser usados?
o Para el sistema actual que está establecido o
o Para un sistema análogo si el sistema aún no está establecido
Detectabilidad
Es una medida de la probabilidad que el evento sea detectado si ocurre. ¿Será detectado?
- ¿Qué métodos están en el lugar disponibles para la detección si el evento ha ocurrido?
- ¿Cuál es la probabilidad de este método de detectar el evento?
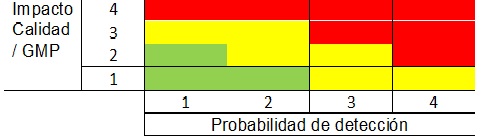
Luego con estos tres scores se calcula el RPN o Risk Priority Number, que es el producto de los 3 puntajes anteriores.
RPN = S x P x D
- A > RPN > Riesgo
- Para los scores de RPN que están en el mismo nivel de riesgo, el score de Severidad le da mayor peso, luego el de Probabilidad y finalmente el de Detectabilidad.
Mitigaciones
Los scores de Severidad (impacto al paciente), raramente pueden ser cambiados, pero hay instancias donde el cambio del material / proceso puede disminuir el impacto al paciente.
Prioridades de mitigación:
Las mitigaciones que reducen la probabilidad deberían ser siempre precedentes a las que mejoran la detectabilidad de la falla (es mejor reducir la probabilidad de que el evento ocurra que depender del método de detección para identificar el problema).
Si la probabilidad no puede ser reducida a nivel aceptable, entonces el método de detección que puede identificar que los eventos han ocurrido deberían ser lo más cercanos / próximos al punto de falla como sea posible
Métodos de detección de ingeniería (ejemplo sistemas de visión, alarmas, etc.), son preferidos sobre aquellos procedimentales como (muestreo AQL, testeos sobre el proceso, etc.). Las mitigaciones procedimentales son las medidas de mitigación más débiles, generalmente recaen sobre el muestreo o testeo para detectar la falla, lo cual probablemente aumenta el impacto del evento. En este caso el producto ya está hecho y el defecto puede impactar de una forma la calidad antes que sea detectado.
Documentación del FMEA
Utilizar la planilla Excel adjunta, en la misma encontrará un ejemplo para ver el uso de la misma. Además contiene notas aclaratorias sobre los campos.
Se establecen para el RPN, rango de valores para clasificar a los riesgos como Bajos, Medio o Altos (la planilla tiene una tabla con criterios, los mismos pueden ser ajustados, pudiendo uno ser más o menos exigente).
Finalmente para todos aquellos riesgos de scores alto (Alto riesgo), deben ser tomadas acciones de mitigación adicionales para reducir el nivel de riesgo a nivel bajo o medio al menos.
Luego de planeadas las acciones de mitigación adicional, el factor de RPN debe ser recalculado de acuerdo a la nueva situación.
Las acciones de mitigación (CAPAs) son seguidas usualmente a través del sistema CAPA del laboratorio y además es vital la verificación de la efectividad de las mismas.
NOTA: es sumamente importante a la hora de estimar la probabilidad de ocurrencia de la falla, conocer las posibles causas de la misma (conocimiento del proceso), de la misma forma que para la determinación de las acciones de mitigación.