Introducción
Cualquier planta GMP que elabore, envase, controle y/o distribuya productos farmacéuticos se apoyará en mayor o menor grado en sistemas computarizados. Estos son fundamentales para garantizar que los procesos y los datos son fiables y seguros. Con el fin de lograr esto, los sistemas computarizados deben mantenerse en un estado validado.
La validación de un sistema computarizado es el establecimiento de pruebas documentadas que proporciona un alto grado de seguridad que funcionará consistentemente, de acuerdo con las especificaciones predeterminadas y atributos de calidad durante todo su ciclo de vida.

¿Qué es una auditoría de sistemas informáticos en una planta GMP?
Una auditoría de los sistemas informáticos es una revisión o inspección de las prácticas, los procedimientos, métodos y normas de la planta GMP que se aplican durante el ciclo de vida del sistema informatizado.
Su objetivo es determinar si la validación del sistema informático es adecuada para satisfacer las expectativas de la regulación y del laboratorio y que el proceso se ejecuta constantemente y de acuerdo con los procedimientos establecidos.
La auditoría también determina cómo se desarrollan, mantienen y utilizan en la Planta los sistemas informáticos.
Gestión de los Sistemas Computarizados
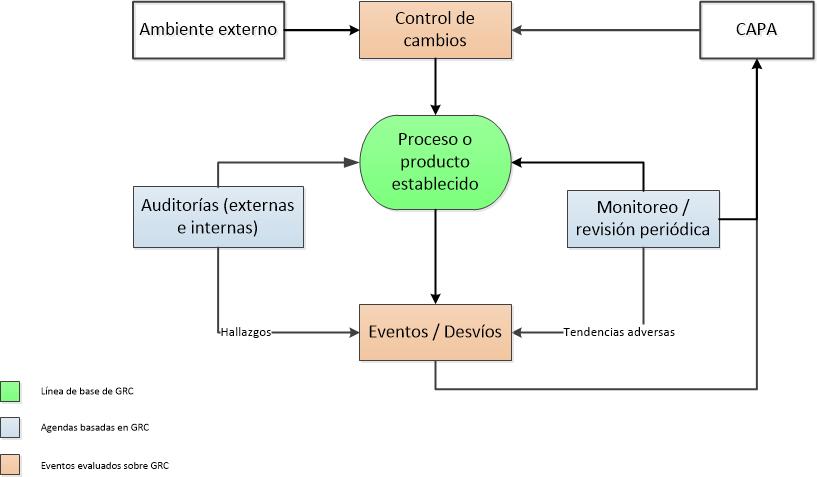
Es importante establecer que los sistemas informáticos son manejados adecuadamente dentro de la planta. Ellos deben estar cubiertos por un sistema de gestión de calidad. Este puede ser el mismo que el resto de la planta o puede ser uno específico, a medida, para los sistemas computarizados.
Procedimientos y normas deben existir para abordar las siguientes áreas:
- Ciclo de Vida de los sistemas computarizados
- Requerimientos funcionales
- Especificaciones de Diseño
- Testeos
- Instalación / Implementación
- Soporte / Mantenimiento
- Control de Cambios / Gestión de la Configuración
- Back UP / Restauración
- Continuidad del negocio y recuperación de desastres
- Seguridad
- Decomiso
- Gestión de desvíos
Es importante determinar el papel de Aseguramiento de la Calidad (QA) en la gestión de los sistemas informáticos. Aseguramiento de la Calidad puede estar involucrado en cada paso del proceso de validación o limitada a la aprobación independiente de las entregas de validación. Es posible que una función separada de Gestión de Calidad IS (ISQM) (dentro de la organización IS) lleve a cabo todas o algunas de las actividades de control de calidad. Es importante que la relación y el papel de las organizaciones de control de calidad e ISQM están documentadas y que la independencia del grupo de desarrollo del sistema se mantenga.
Los procedimientos deben describir qué nivel de aprobación se requiere para cada entregable de validación. Como mínimo, la aprobación QA se esperaría para los Planes de validación, requerimientos e informes de validación y documentación de control de cambios. Si se producen desviaciones a los procedimientos, confirmar que están documentados, evaluados y aprobados.
Un inventario de los sistemas debe existir para identificar todos los sistemas de la planta, detallando el propietario y el impacto GMP. Cualquier sistema con un impacto GMP que se encuentra actualmente en funcionamiento debe ser validado. La infraestructura de IT también debe ser identificada.
Sistemas críticos GMP pueden incluir:
- Sistema de Control de Procesos (SCADA (Supervisory control and data acquisition), autoclaves)
- Sistemas de Monitoreo Ambiental
- LIMS (Laboratory Information Management System)
- MES (Manufacturing Execution Systems)
- Sistema de gestión de documentos
- ERP (Enterprise Resource Planning) por ejemplo, BPCS, SAP, etc.
Debe quedar claro cómo se determina, evalúa y documenta el impacto GMP o criticidad de un sistema.
Validación
Entregables de validación
En general, las prácticas de documentación GMP estándar debe aplicarse a toda la documentación de validación del sistema computarizado. Como mínimo esto incluye paginado, datos, historial de revisiones, versión de los documentos, aprobaciones, y otras buenas prácticas de documentación.
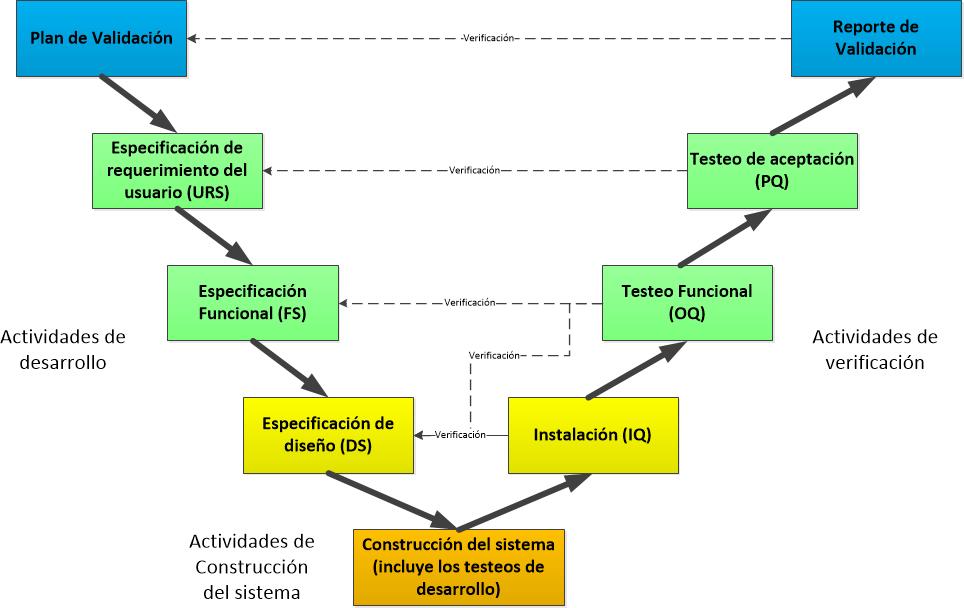
Plan de Validación
El Plan de Validación proporcionará información de planificación específico en lo que respecta a las actividades de validación que se realizarán para el sistema. El Plan de Validación describe el enfoque de validación que debe tomarse, indica los entregables de validación y proporciona criterios de aceptación claros. El alcance y la profundidad de validación del sistema computarizado varían, dependiendo de la naturaleza y la complejidad del sistema. Un enfoque de categorización se puede tomar donde los sistemas se agrupan en función de su complejidad y su uso.
La GAMP5 clasifica a los sistemas en 4 categorías:
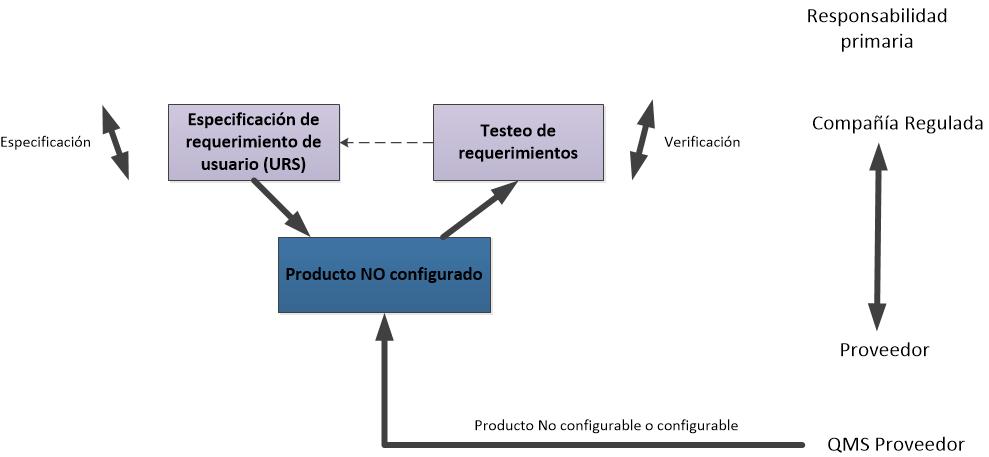
- 1. Sistemas Operativos disponibles en el mercado. Las aplicaciones se desarrollan para funcionar bajo el control de estos sistemas operativos. Ej. DOS. Otros sistemas de categoría 1 son los Firmware, los instrumentos y controladores a menudo incorporan firmware. La configuración puede ser necesaria para el set up y los parámetros del proceso del entorno de ejecución.
- 3. Paquetes de software estándar
Son paquetes estándares disponibles en el mercado, proporcionando una solución de estante. Se requiere configuración limitada para establecer el entorno de ejecución.
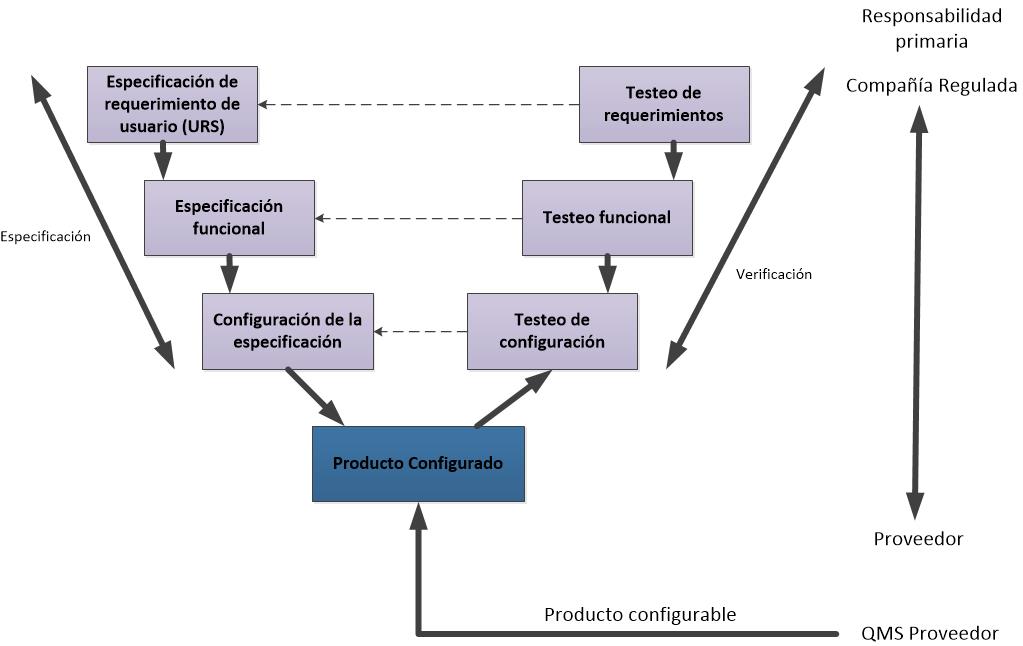
- 4. Paquetes de Software configurables
Los paquetes de software configurables que proporcionan interfaces y funciones que permiten la configuración de usuario o procesos de negocio específicos estándar.
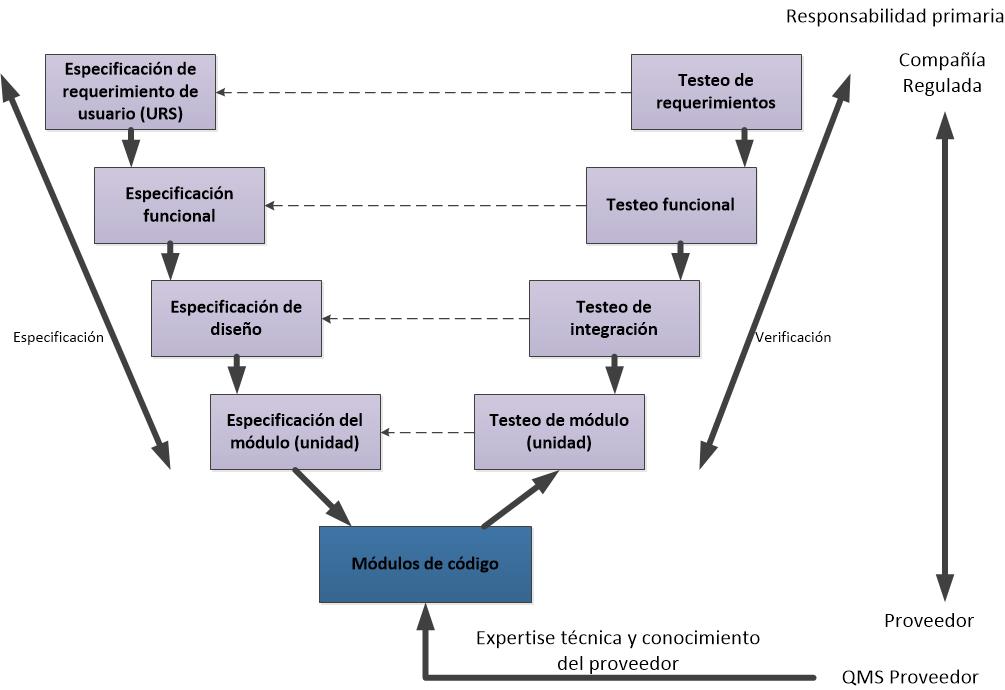
- 5. Software a medida
Software desarrollado para satisfacer las necesidades específicas del negocio.
El plan debe indicar la aprobación del propietario de procesos de negocio o el propietario del sistema, los representantes técnicos y Aseguramiento de la Calidad.
Especificación de requerimientos
La especificación de requisitos (puede ser llamada como URS – especificación de requerimientos de usuarios o simplemente requisitos del usuario) describe lo que el usuario necesita que haga el sistema.
Los requisitos deben ser únicos y priorizados. Deben estar escritos a un nivel detallado, identificando con precisión los criterios aceptables para el éxito desde la perspectiva de los usuarios.
Especificación funcional
La especificación funcional (puede ser llamada como Especificaciones de Diseño) describe cómo el sistema está diseñado para alcanzar los requisitos funcionales. Este entregable es un documento técnico que identifica la solución técnica para la aplicación, el software subyacente y el hardware necesario para apoyar el sistema. Debe haber trazabilidad clara entre los requisitos y el diseño funcional. Esto puede conseguirse usando una matriz, referencias cruzadas, la numeración común o cualquier otro enfoque que hace que sea claro cómo cada requisito se satisface mediante el diseño.
Programación del sistema.
Codificación deben cumplir con los estándares de programación para las pantallas, menús, anotaciones de código, etc. Cuando sea posible utilizar estándares de codificación de la industria, los mismos deben ser usados. El cumplimiento de los estándares de codificación debe ser evaluado a través de la revisión formal de código. Un enfoque basado en el riesgo se puede aplicar a la revisión de código con el código seleccionado basa en la complejidad, criticidad o la experiencia del desarrollador.
Los defectos encontrados durante la revisión de código deben ser registrados con las acciones correctivas y preventivas, según corresponda.
Procedimientos para el mantenimiento y el control de versiones múltiples de código fuente deben estar disponibles. En la práctica esto se logra a menudo usando software de gestión de la configuración comercial por ejemplo, CVS, Microsoft Visual SourceSafe o IBM Rational ClearQuest.
Siempre que sea posible deben ser utilizados ambientes separados, para el desarrollo y para los testeos. Estos deben ser lo más similar posible al entorno de producción para el sistema con el fin de asegurar la validez de las pruebas del sistema.
Especificación de testeos
La especificación de testeos describe las pruebas que se realizan para asegurar que el sistema cumple con los requisitos de los usuarios. Los scripts de testeos deben ser escritos de forma clara. Las pruebas deben probar límites, las fallas y condiciones de estrés, así como la ejecución exitosa de la funcionalidad requerida. Debe haber trazabilidad de los requisitos. Debe ser posible demostrar que toda la funcionalidad requerida ha sido probada de forma adecuada.
Es una buena práctica para los tests para ser escritos por alguien que no sea el desarrollador y ejecutados por otra persona independiente.
Resultados esperados deben estar claramente identificados y los resultados obtenidos se deben documentar con pruebas suficientes para demostrar el cumplimiento. Las desviaciones de las etapas de testeso o resultados esperados deben estar documentados.
Las fallas de los tests deben ser registradas e investigadas. Dependiendo del impacto de la falla y la criticidad de las opciones de la función, puede incluir corregir el código y volver a probar la función (y otras funciones implicadas) o proporcionar una solución de procedimiento.
Las pruebas pueden llevarse a cabo utilizando un software de testeos automatizados. Deben existir procedimientos que cubran el mantenimiento de la herramienta de testeos y los scripts de testeos. Los softwares y herramientas de testeos automatizadas deben ser validados.
Las pruebas pueden existir en diferentes niveles con:
- Testeos de unidad (o módulo) – se trata de pruebas técnicas, a veces realizada por el desarrollador, para demostrar que los módulos de código individual o funciones performan correctamente
- Testeos de integración – aseguran que los módulos funcionan correctamente juntos como un sistema en su conjunto. Esta fase puede incluir la verificación de las interfaces con otros sistemas, aunque las pruebas de la interfaz puede ser una fase separada.
- Testeos de aceptación de usuario – esta fase confirma que las necesidades de los usuarios se han cumplido
Es aceptable tener cada vez mayor formalidad aplicada a fases de prueba en términos del grado de documentación de la prueba y las pruebas recogidas.
Informe de validación
El informe de validación resume las actividades de validación asociadas con el desarrollo del sistema. Contiene un resumen de las conclusiones de testeos e informes sobre otros requisitos de validación, por ejemplo, entrenamiento de usuarios y procedimientos actualizados. Proporciona recomendaciones para la aceptación del sistema. El Informe también presenta una estrategia para mantener el sistema en un estado validado. Cuando hay alguna excepción a los entregables requeridos por el plan de validación, éstas se detallan en el informe junto con una justificación. Cualquier acción a ser completada después de la implementación del sistema debe ser rastreada hasta su finalización.
El Informe de Validación debe ser aprobado antes de que el sistema entre en uso operacional.
Liberación / instalación
La liberación e implementación del sistema se basa en el cumplimiento de los entregables de validación y otros requisitos previos necesarios para la gestión del sistema. Estos pueden incluir: una construcción confirmada desde el sistema de gestión de configuración, los procedimientos de instalación, manuales de referencia técnica, manuales de referencia del sistema, manuales de operación del sistema, procedimientos de procesos, capacitación.
En el artículo II, desarrollamos, estrategia de mantenimiento y gestión del cambio, medidas de seguridad, firmas y registros electrónicos, e integridad de datos. para leerlo haga click aquí.
Además para aquellos interesados en el tema, le proponemos un Taller sobre el tema, consúltenos en info@cgmpdoc.com

{kind=link}
{kind=link}