Hay dos componentes necesarios para que una auditoría sea exitosa.
El primero es un auditor con las habilidades, la educación y la experiencia correcta o necesaria (ver haciendo click aquí).
El segundo es el proceso de auditoría por sí mismo.
El proceso de auditoría puede comenzar algunos meses antes de que la auditoría se lleve a cabo. Las etapas del proceso de auditoría son:
Preparación de la auditoría
- Determinar quién conducirá la auditoría – un individuo o un equipo.
En el caso de un equipo, el auditor líder será responsable de asegurar que la auditoría es manejada efectivamente y eficientemente.
- Desarrollar la agenda de la auditoría
Para conducir una auditoría eficiente y efectiva, debe ser elaborada una agenda. Esto ayudará a mantener el flujo de la misma sin problemas y hará mejor el tiempo del auditado y del auditor.
El propósito de la auditoría debería estar claro para ambos, el auditado y el auditor. Puesto que efectuará una auditoría GMP, Ud. Debería considerar los siguientes temas GMP:
- Sistemas de Calidad
- Gestión de materiales
- Controles de calidad del laboratorio
- Sistemas de producción
- Instalaciones y equipos
- Empaque y rotulado
Durante la auditoría el auditor debe planear prestar especial atención a los sistemas de calidad, la forma en la cual el laboratorio maneja los desvíos, los reclamos y los resultados de los ensayos OOS (fuera de especificaciones), esto le dará una visión de la cultura de calidad de la compañía y su adherencia a las cGMPs.
- Confirmar la auditoría y acordar una fecha con el proveedor /área a ser auditada.
Trabajar con el auditado para determinar un tiempo para ajustarse dentro del marco de tiempo que sea conveniente para ambas partes.
- Revisar documentos, incluir documentos recibidos desde la empresa o el laboratorio
- Preparar notas para la auditoría
Luego de revisar la información de soporte y los documentos de referencia y los estándares, el auditor prepara notas que lo ayudará a efectuar la auditoría, así como una lista de documentos a revisar.
Potenciales actividades de la auditoría en el laboratorio
Todas las auditorías deben ser conducidas de una forma abierta, el auditor debe discutir los hallazgos y expectativas con el auditado mientras conduce la auditoría. La auditoría es una oportunidad de identificar oportunidades de mejora para el auditado.
- Conducir una reunión de apertura
Al arribar al laboratorio, el auditor / equipo auditor debe efectuar una reunión de apertura con los responsables del laboratorio (QA, producción, dirección, otros representantes asignados). Durante la reunión de apertura, el auditor debe asegurarse que el auditado entienda su rol en la auditoría.
- Recorrer los depósitos, instalaciones de manufactura y laboratorios, si es apropiado.
Las auditorías pueden comenzar con una recorrida a las instalaciones y al laboratorio, es recomendable “seguir un producto” (desde la recepción de materiales hasta la liberación del producto terminado).
Durante la recorrida, verificar que los SOPs e instrucciones están siendo seguidos y los registros se completan con exactitud.
A medida que realiza la auditoría el auditor puede seguir las notas preparatorias, para asegurar que cubre la auditoría completa.
Las observaciones de la auditoría pueden ser clasificadas en críticas, mayores y menores.
Si un tema o desvío crítico es observado, el mismo debe ser comunicado inmediatamente al management del laboratorio y al responsable auditado
- Entrevistar analistas y operadores (si es necesario).
- Efectuar una reunión informativa de la auditoría con el auditado, si es apropiado.
Para las revisiones informativas donde las observaciones y hallazgos deberían ser revisadas con el auditado como también las áreas de preocupación para asegurar que no habrá sorpresas o malos entendidos en el cierre final de la auditoría.
La agenda de la auditoría puede ser actualizada si son requeridos cambios basados en las observaciones o recursos.
- Efectuar reuniones de auditoría con el equipo, si un enfoque de equipo auditor es utilizado.
Estas reuniones son mantenidas para ayudar al equipo auditor a mantenerse enfocado en el objetivo de la auditoría y la auditoría en curso.
- Revisar la documentación del auditado
Esto puede incluir SOPs, documentación de manufactura y de ensayos, datos crudos asociados, protocolos de calificación y validación, y reportes de investigaciones.
- Conducir la reunión de cierre
La reunión de cierre es el foro usado para presentar verbalmente los resultados de la auditoría al management auditado. Es esencial que todos los participantes claves de la auditoría y el management del auditado participen de la reunión de cierre.
Seguimiento de las actividades de la auditoría
- Documentar las observaciones en un reporte formal
Los mismos están basados en las anotaciones que el auditor tomó durante la auditoría. Los mismos pueden ser usados por diferentes personas para diferentes motivos, por eso necesitan ser claros y escritos de una forma consistente como también contener la información relevante y necesaria.
Los reportes de auditoría deben ser normalmente emitidos dentro de un cierto plazo, así como también las respuestas conteniendo el plan de acción.
- Evaluar las respuestas recibidas desde la empresa / laboratorio
Luego que las respuestas son recibidas, es necesario analizarlas y determinar si las acciones correctivas planteadas y los riesgos propuestos son aceptables. Una vez recibida la respuesta, y si las respuestas son consideradas aceptables, el auditor tiene la responsabilidad de responder dentro de un período de tiempo definido.
- Cerrar la auditoría
- Efectuar el seguimiento de los CAPAs generados
El seguimiento puede requerir nuevas visitas al laboratorio o la recepción de las evidencias de las acciones cumplidas, para darle cierre a la acción pendiente.
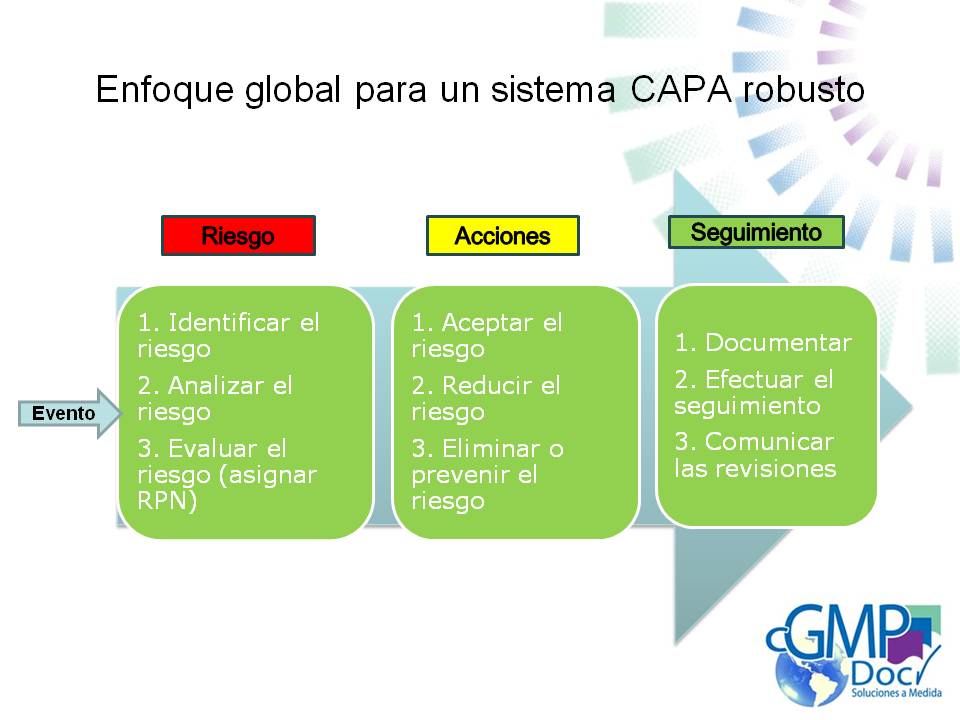
Evaluación de riesgo durante el proceso de auditoría:
El auditor toma decisiones basadas en el riesgo a través del proceso de auditorías, por ejemplo dado que No es posible cubrir todo durante el proceso de auditoría, es necesario evaluar los riesgos, tal vez efectuar un análisis de riesgo formal y hacer un juicio sobre que cubrir. Este juicio que necesita hacer puede necesitar cambiar durante el proceso de auditoría basado en nuevos conocimientos.
Herramientas de análisis de riesgos:
Para soportar la evaluación de riesgo de un proveedor, el auditor debe elegir usar una herramienta de análisis de riesgo. Una de las herramientas formales de análisis de riesgo usadas por los auditores es Failure Modes Effects Analysis (FMEA).
Si Ud. está interesado en entrenar a su equipo auditor, consulte en info@cgmpdoc.com. Podemos ofrecerle materiales de entrenamiento para Auditores, check lists para que Ud. Efectúe las distintas auditorías y además tenemos la posibilidad de entrenar a su equipo “In Company”.