¿Has oído hablar de bambú? Bueno los que practicamos judo y tuvimos la oportunidad de ser discipulos de maestros japoneses, si.

La historia que solían contarnos era la siguiente:
Si siembras una semilla de bambú en el suelo y le das el agua suficiente durante todo el verano obtendrás … nada.
Si luego lo continuas regando y le añades un poco de fertilizante en el verano siguiente obtendrás … nada.
Si le das un poco de amor y cuidado durante el tercer verano obtendrás … nada.
Se paciente durante un verano más y obtendrás … nada.
PERO … en el quinto verano, el brote de bambú deseado y muy ausente, aparecerá y crecerá a su altura máxima a unas pocas semanas … tan alto como 30 m en un verano.
Esa es la recompensa vale la pena esperar!
Pero hablemos algo sobre el bambú…
El bambú (de la familia de las gramineas) es una de las plantas de más rápido crecimiento en el mundo, puede crecer tan alto como 2.5 m en un día!
Constituye un gran símbolo oriental y tiene mucha presencia en nuestra decoración occidental actual, pero en realidad lo que nosotros utilizamos para decorar nuestras casas, como si fuese bambú, no es bambú, sino drácena sanderiana, aunque realmente son difíciles de distinguir.
Se utiliza como un material para la construcción, la escritura, pero también puede ser utilizado como un arma o un instrumento musical. El bambú se utiliza en todo el mundo, pero no crece tan rápido en todas partes. Necesita un clima y un suelo adecuado. El bambú no puede crecer hasta que el sistema rizoma que la sustenta es extensa y bien cuidada, sin raíces no habrá arbol.
No puedo dejar de hacer un paralelo con la forma en que ponemos algunos esfuerzos en la construcción de una cultura de calidad en nuestras organizaciones.
¿Regamos con paciencia a nuestros futuros brotes durante cinco años antes de esperar resultados? Generalmente no.
¿Vigilamos atentamente el crecimiento del sistema de rizomas, las raíces de nuestra cultura para asegurar con el tiempo el magnífico tronco que producirá? Generalmente No.
¿Creemos que tendremos éxito después de años de esfuerzos, tenemos fe en nuestras raíces? Ummm…no
¿Queremos resultados y rápido? Claro que sí.
Pero, ¿realmente pensamos que podemos lograr esto sin un sistema de raíces fuertes y eficientes? Realmente no podemos creer eso.
Hace poco tiempo tuve la oportunidad de participar en una reunión en la cual participaban dos profesionales de la industria farmacéutica, jóvenes profesionales que están haciendo una excelente carrera en un prestigioso laboratorio multinacional.
Y en un momento quedé estupefacto, cuando uno le decía al otro:
“Con el fin de llamar la atención de la Dirección, TENEMOS que hablar de la cultura de calidad y de cómo cambiarla de forma rápida»
La imagen del bambú vino a mi mente, pero el significado de ese ”de forma rápida” me generaba dudas.
Finalmente cuando dijo: “… Dentro de 6 a 12 meses. » les pedí la oportunidad de comentarles mi opinión, tratando de argumentar a favor de un tiempo, y les dije:
“Ustedes no tienen que hacer que suceda por completo en 12 meses, pero pueden mostrar muchos resultados en ese lapso de tiempo, y luego les muestran como sostenerlos y aumentarlos con el tiempo… »
Todos sabemos y entendemos que un cambio cultural profundo no puede suceder durante la noche, pero podemos confiadamente esperar resultados rápidos en un corto espacio de tiempo.
Pero me permití decirles además que para ello necesitamos:
- Una clara visión de la “cultura” de Calidad que deseamos
- Una clara señal de forma continua a partir de los líderes de las organizaciones
- Objetivos claros
- Compromiso real de todos
Creo que ese es el camino correcto, pero, les parece que vamos muy lento?
Napoleón diría: “Vísteme despacio que estoy apurado..».
Para los interesados en los sistemas de calidad, quiero compartirles como los sistemas de calidad están integrados a la gestión de riesgo de calidad, en un simple flujo de proceso, solo tienen que enviarnos un email a info@cgmpdoc.com y con gusto se lo enviaremos.
Soporte, estrategia de mantenimiento y Gestión del Cambio
Con el fin de mantener el estado validado, el sistema debe ser soportado y mantenido y los cambios deben ser gestionados adecuadamente. Un mecanismo debe estar en su lugar para registrar los problemas del sistema, investigarlos y seguirlos hasta el final con el seguimiento apropiado de las acciones correctivas y preventivas, según corresponda.
Los cambios en el sistema pueden ser consecuencia de problemas o cambios en los procesos de negocio y el uso del sistema. El impacto de cualquier cambio propuesto en la funcionalidad existente debe ser evaluado antes de su aprobación. Los testeos de soporte del cambio deben demostrar que
(a) la funcionalidad cambiada performa como se esperaba y
(b) no hay un impacto inesperado en las funciones del sistema relacionadas.
De forma periódica debe haber una revisión de la gestión del sistema para determinar que necesita hacerse para mantener el estado validado. Inputs a la revisión de la gestión pueden incluir:
- Cantidad de cambios del sistema
- Problemas comunes de los usuarios
- Cambios técnicos a la infraestructura de apoyo al sistema
- Soporte continuo a las partes de origen comercial del sistema (hardware o software)
- Cambios en el uso del sistema
- Cambios en los procesos de negocio soportados por el sistema
Las recomendaciones resultantes de la revisión periódica deben ser registrados formalmente y las acciones deben ser seguidas.
Siempre que sea posible, el sistema debe ser mantenido con los parches de seguridad actuales de los sistemas operativos, bases de datos y software de aplicación con el fin de proteger el proceso y los datos del acceso y cambio no autorizado (hacking).
Cuando al sistema se puede acceder directamente por medio del proveedor de software (por acceso telefónico o Internet) este acceso debe ser controlado de manera que los cambios realizados sean gestionados de forma normal.
Copias de seguridad y restauración
Con el fin de asegurar el sistema y sus datos, deben ser identificados requerimientos de back up y recuperación para todos los sistemas que tienen impacto sobre las GMP. La frecuencia con que se efectúan las copias de seguridad debe ser determinada de acuerdo a la criticidad del sistema. Los datos respaldados deben estar bien seguros y la recuperación de los mismos debe ser probada periódicamente, para asegurar que es exitosa.
Continuidad de Negocio / Recuperación de Desastres
La pérdida de sistemas informáticos puede ocurrir por una variedad de razones, que van desde fallas en los equipos, de red o fallas de software por una catástrofe en la planta. Se prevé que la mayoría de los problemas de sistemas computarizados o infraestructuras se resolverán rápidamente por procesos de apoyo existentes para minimizar el impacto en los usuarios finales del sistema. Un problema serio puede, sin embargo, convertirse en un desastre si se deja sin resolver.
El período inmediato que rodea a un desastre puede dar lugar a confusión, el caos y la interrupción significativa de la operación del negocio de rutina. Las medidas adoptadas en las primeras horas de una crisis tienen una incidencia significativa en la severidad y la velocidad máxima para reanudar las operaciones normales del negocio. La creación de planes estructurados ayudará proporcionando responsabilidades y acciones claramente definidas. Estos procedimientos, junto con un poco de preparación antes del desastre, asegurarán que el impacto en el negocio de cualquier desastre puede ser gestionado y minimizado.
La continuidad del negocio (BC) y la recuperación de desastres (DR) son dos procesos paralelos que se preparan antes del desastre y se ejecutan después de que ocurre el desastre. Planificación BC describe cómo los procesos de negocio y actividades continúan en ausencia de los sistemas informáticos de apoyo. La planificación del BC depende enteramente de la criticidad del sistema. Para los sistemas la planificación BC puede ser la de no hacer nada hasta que se restablezca el sistema.
La planificación del DR describe cómo los sistemas se restauran a un estado operativo (incluidos los datos) y deben estar en su lugar para todos los sistemas computarizados e infraestructura. Una vez más un enfoque basado en el riesgo se toma a menudo.
Para asegurar que los planes son eficaces en el caso de un desastre tienen que ser precisos, oportunos y completos. Ellos deben ser revisados y actualizados periódicamente. Cuando sea práctico la revisión debe incluir una prueba o ejercicio de los planes.
Medidas de seguridad
Las medidas de seguridad deben estar en su lugar para evitar el acceso no autorizado al sistema. La seguridad física puede implicar el control de acceso al centro de datos o restringir el acceso a áreas específicas dentro de la instalación, por ejemplo los laboratorios pertinentes o áreas de la planta. Acceso lógico puede incluir controles sobre el acceso al software del sistema, lo que restringe las funciones críticas del sistema para los usuarios especificados y cambiar las contraseñas en los sistemas operativos, bases de datos y software de aplicación. Una consideración importante es mantener la seguridad de los componentes del sistema actualizado con los parches de seguridad proporcionados por el vendedor. El objetivo de la seguridad física y lógica es la de proteger la funcionalidad y los datos del sistema del cambio no autorizado.
Registros Electrónicos y Firmas Electrónicas
Cuando hay sistemas computarizados de apoyo a los procesos críticos de GMP en una instalación y gran parte de los datos crudos (raw data) mantenidos electrónicamente, es importante que los registros y firmas asociadas son seguras, confiables y atribuible. Es importante que el personal de la planta entienda que sus firmas electrónicas son el equivalente de la firma manuscrita. El Audit trail debe registrar todas los detalles necesarios incluyendo quién creó o modificó un registro y cuándo (21 CFR Parte 11).
Un par de temas más…
Retiro o decomiso del sistema
Procedimiento mediante la cual un sistema es retirado del uso productivo, aquí el código se archiva, los datos son convertidos o migrados (si aplica), y el sistema está desmontado y colocado fuera de servicio. El enfoque utilizado para el retiro sistema debe mantener la integridad de los datos o la necesidad de la utilización de los datos en el futuro.
Migración de datos
La migración de datos es un proceso de una sola vez donde los datos se mueven de un sistema a otro sistema. Esto puede ser debido a una actualización del sistema o debido a la sustitución de un sistema existente con un nuevo sistema. Este no es un proceso continuo entre dos sistemas.
El enfoque de la migración de datos debe consistir en un plan de migración, la identificación de los datos que se migra, el proceso real de la migración de los datos, y un proceso de verificación de la migración para determinar que el proceso de migración era precisa y exitosa.
Las desviaciones del proceso de migración de datos deben ser documentos y resueltas de acuerdo a su criticidad.
Espero que les resulte interesante, a los que ingresaron por acá, les dejo el link al artículo I, haciendo click aquí.
Introducción
Cualquier planta GMP que elabore, envase, controle y/o distribuya productos farmacéuticos se apoyará en mayor o menor grado en sistemas computarizados. Estos son fundamentales para garantizar que los procesos y los datos son fiables y seguros. Con el fin de lograr esto, los sistemas computarizados deben mantenerse en un estado validado.
La validación de un sistema computarizado es el establecimiento de pruebas documentadas que proporciona un alto grado de seguridad que funcionará consistentemente, de acuerdo con las especificaciones predeterminadas y atributos de calidad durante todo su ciclo de vida.

¿Qué es una auditoría de sistemas informáticos en una planta GMP?
Una auditoría de los sistemas informáticos es una revisión o inspección de las prácticas, los procedimientos, métodos y normas de la planta GMP que se aplican durante el ciclo de vida del sistema informatizado.
Su objetivo es determinar si la validación del sistema informático es adecuada para satisfacer las expectativas de la regulación y del laboratorio y que el proceso se ejecuta constantemente y de acuerdo con los procedimientos establecidos.
La auditoría también determina cómo se desarrollan, mantienen y utilizan en la Planta los sistemas informáticos.
Gestión de los Sistemas Computarizados
Es importante establecer que los sistemas informáticos son manejados adecuadamente dentro de la planta. Ellos deben estar cubiertos por un sistema de gestión de calidad. Este puede ser el mismo que el resto de la planta o puede ser uno específico, a medida, para los sistemas computarizados.
Procedimientos y normas deben existir para abordar las siguientes áreas:
- Ciclo de Vida de los sistemas computarizados
- Requerimientos funcionales
- Especificaciones de Diseño
- Testeos
- Instalación / Implementación
- Soporte / Mantenimiento
- Control de Cambios / Gestión de la Configuración
- Back UP / Restauración
- Continuidad del negocio y recuperación de desastres
- Seguridad
- Decomiso
- Gestión de desvíos
Es importante determinar el papel de Aseguramiento de la Calidad (QA) en la gestión de los sistemas informáticos. Aseguramiento de la Calidad puede estar involucrado en cada paso del proceso de validación o limitada a la aprobación independiente de las entregas de validación. Es posible que una función separada de Gestión de Calidad IS (ISQM) (dentro de la organización IS) lleve a cabo todas o algunas de las actividades de control de calidad. Es importante que la relación y el papel de las organizaciones de control de calidad e ISQM están documentadas y que la independencia del grupo de desarrollo del sistema se mantenga.
Los procedimientos deben describir qué nivel de aprobación se requiere para cada entregable de validación. Como mínimo, la aprobación QA se esperaría para los Planes de validación, requerimientos e informes de validación y documentación de control de cambios. Si se producen desviaciones a los procedimientos, confirmar que están documentados, evaluados y aprobados.
Un inventario de los sistemas debe existir para identificar todos los sistemas de la planta, detallando el propietario y el impacto GMP. Cualquier sistema con un impacto GMP que se encuentra actualmente en funcionamiento debe ser validado. La infraestructura de IT también debe ser identificada.
Sistemas críticos GMP pueden incluir:
- Sistema de Control de Procesos (SCADA (Supervisory control and data acquisition), autoclaves)
- Sistemas de Monitoreo Ambiental
- LIMS (Laboratory Information Management System)
- MES (Manufacturing Execution Systems)
- Sistema de gestión de documentos
- ERP (Enterprise Resource Planning) por ejemplo, BPCS, SAP, etc.
Debe quedar claro cómo se determina, evalúa y documenta el impacto GMP o criticidad de un sistema.
Validación
Entregables de validación
En general, las prácticas de documentación GMP estándar debe aplicarse a toda la documentación de validación del sistema computarizado. Como mínimo esto incluye paginado, datos, historial de revisiones, versión de los documentos, aprobaciones, y otras buenas prácticas de documentación.
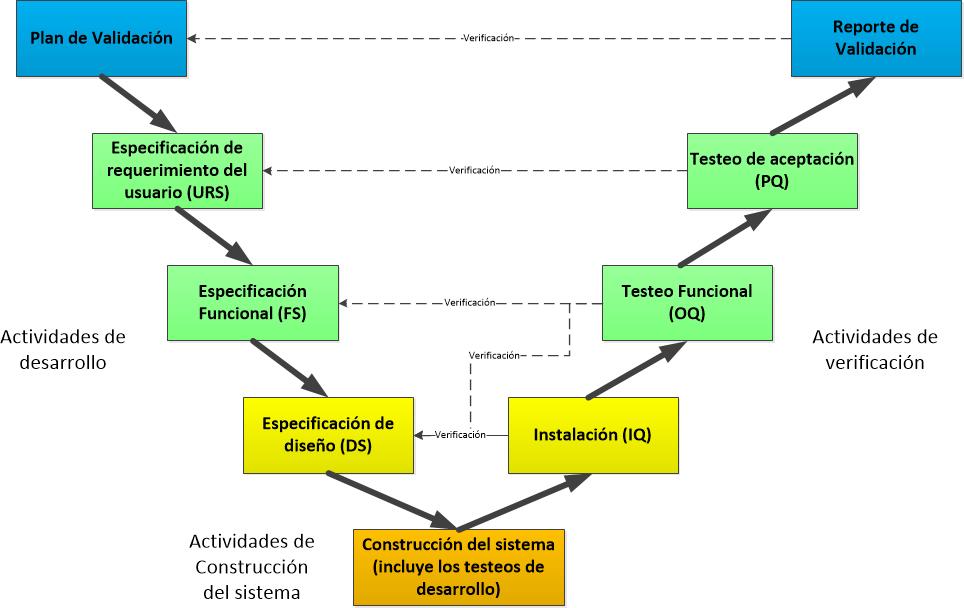
Plan de Validación
El Plan de Validación proporcionará información de planificación específico en lo que respecta a las actividades de validación que se realizarán para el sistema. El Plan de Validación describe el enfoque de validación que debe tomarse, indica los entregables de validación y proporciona criterios de aceptación claros. El alcance y la profundidad de validación del sistema computarizado varían, dependiendo de la naturaleza y la complejidad del sistema. Un enfoque de categorización se puede tomar donde los sistemas se agrupan en función de su complejidad y su uso.
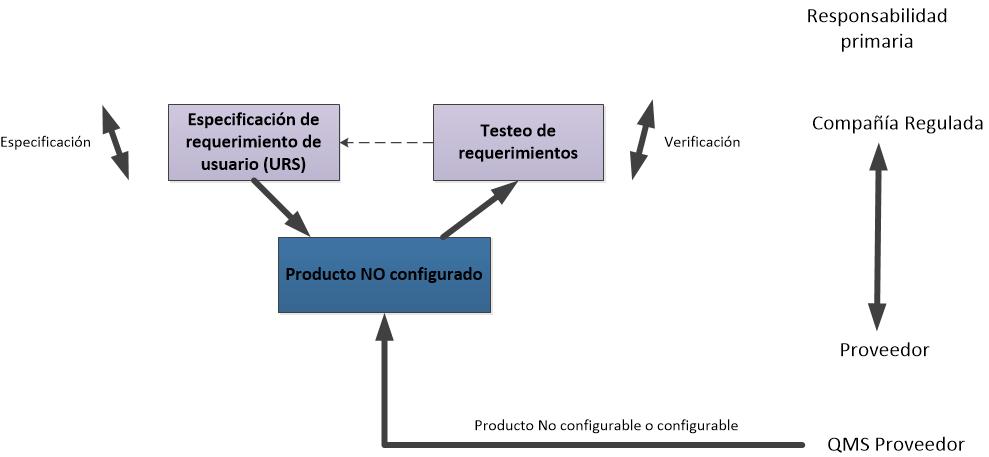
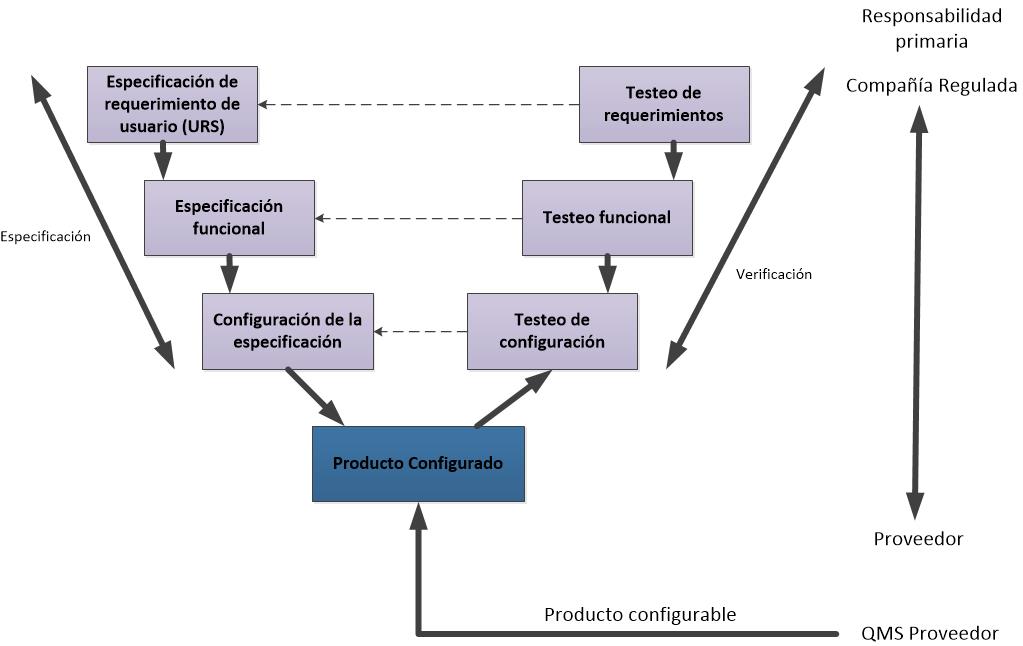
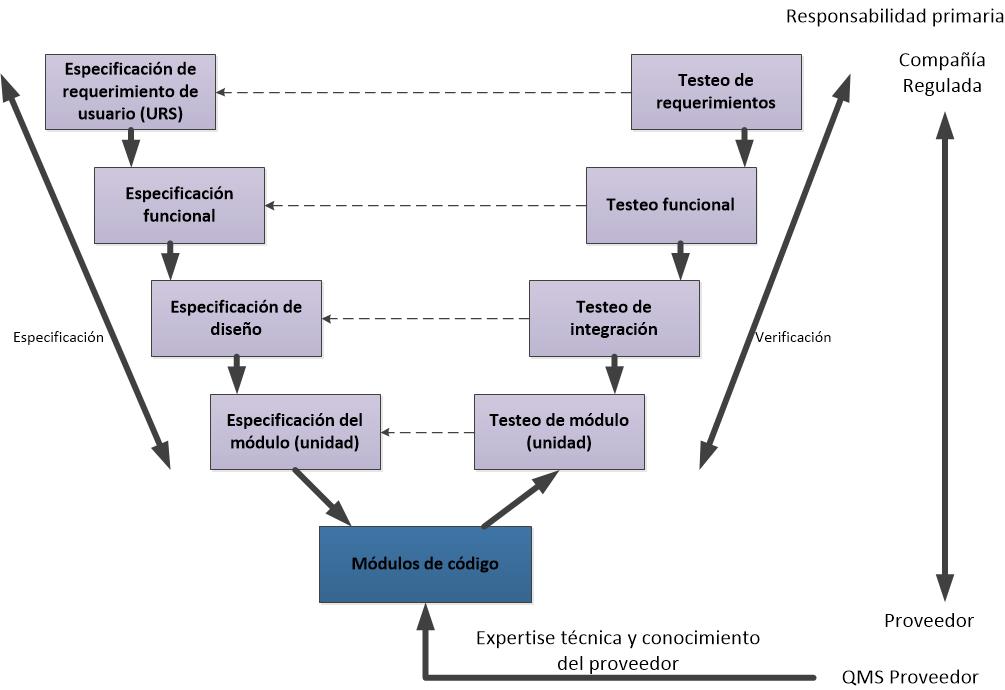
La GAMP5 clasifica a los sistemas en 4 categorías:
- 1. Sistemas Operativos disponibles en el mercado. Las aplicaciones se desarrollan para funcionar bajo el control de estos sistemas operativos. Ej. DOS. Otros sistemas de categoría 1 son los Firmware, los instrumentos y controladores a menudo incorporan firmware. La configuración puede ser necesaria para el set up y los parámetros del proceso del entorno de ejecución.
- 3. Paquetes de software estándar
Son paquetes estándares disponibles en el mercado, proporcionando una solución de estante. Se requiere configuración limitada para establecer el entorno de ejecución. - 4. Paquetes de Software configurables
Los paquetes de software configurables que proporcionan interfaces y funciones que permiten la configuración de usuario o procesos de negocio específicos estándar. - 5. Software a medida
Software desarrollado para satisfacer las necesidades específicas del negocio.
El plan debe indicar la aprobación del propietario de procesos de negocio o el propietario del sistema, los representantes técnicos y Aseguramiento de la Calidad.
Especificación de requerimientos
La especificación de requisitos (puede ser llamada como URS – especificación de requerimientos de usuarios o simplemente requisitos del usuario) describe lo que el usuario necesita que haga el sistema.
Los requisitos deben ser únicos y priorizados. Deben estar escritos a un nivel detallado, identificando con precisión los criterios aceptables para el éxito desde la perspectiva de los usuarios.
Especificación funcional
La especificación funcional (puede ser llamada como Especificaciones de Diseño) describe cómo el sistema está diseñado para alcanzar los requisitos funcionales. Este entregable es un documento técnico que identifica la solución técnica para la aplicación, el software subyacente y el hardware necesario para apoyar el sistema. Debe haber trazabilidad clara entre los requisitos y el diseño funcional. Esto puede conseguirse usando una matriz, referencias cruzadas, la numeración común o cualquier otro enfoque que hace que sea claro cómo cada requisito se satisface mediante el diseño.
Programación del sistema.
Codificación deben cumplir con los estándares de programación para las pantallas, menús, anotaciones de código, etc. Cuando sea posible utilizar estándares de codificación de la industria, los mismos deben ser usados. El cumplimiento de los estándares de codificación debe ser evaluado a través de la revisión formal de código. Un enfoque basado en el riesgo se puede aplicar a la revisión de código con el código seleccionado basa en la complejidad, criticidad o la experiencia del desarrollador.
Los defectos encontrados durante la revisión de código deben ser registrados con las acciones correctivas y preventivas, según corresponda.
Procedimientos para el mantenimiento y el control de versiones múltiples de código fuente deben estar disponibles. En la práctica esto se logra a menudo usando software de gestión de la configuración comercial por ejemplo, CVS, Microsoft Visual SourceSafe o IBM Rational ClearQuest.
Siempre que sea posible deben ser utilizados ambientes separados, para el desarrollo y para los testeos. Estos deben ser lo más similar posible al entorno de producción para el sistema con el fin de asegurar la validez de las pruebas del sistema.
Especificación de testeos
La especificación de testeos describe las pruebas que se realizan para asegurar que el sistema cumple con los requisitos de los usuarios. Los scripts de testeos deben ser escritos de forma clara. Las pruebas deben probar límites, las fallas y condiciones de estrés, así como la ejecución exitosa de la funcionalidad requerida. Debe haber trazabilidad de los requisitos. Debe ser posible demostrar que toda la funcionalidad requerida ha sido probada de forma adecuada.
Es una buena práctica para los tests para ser escritos por alguien que no sea el desarrollador y ejecutados por otra persona independiente.
Resultados esperados deben estar claramente identificados y los resultados obtenidos se deben documentar con pruebas suficientes para demostrar el cumplimiento. Las desviaciones de las etapas de testeso o resultados esperados deben estar documentados.
Las fallas de los tests deben ser registradas e investigadas. Dependiendo del impacto de la falla y la criticidad de las opciones de la función, puede incluir corregir el código y volver a probar la función (y otras funciones implicadas) o proporcionar una solución de procedimiento.
Las pruebas pueden llevarse a cabo utilizando un software de testeos automatizados. Deben existir procedimientos que cubran el mantenimiento de la herramienta de testeos y los scripts de testeos. Los softwares y herramientas de testeos automatizadas deben ser validados.
Las pruebas pueden existir en diferentes niveles con:
- Testeos de unidad (o módulo) – se trata de pruebas técnicas, a veces realizada por el desarrollador, para demostrar que los módulos de código individual o funciones performan correctamente
- Testeos de integración – aseguran que los módulos funcionan correctamente juntos como un sistema en su conjunto. Esta fase puede incluir la verificación de las interfaces con otros sistemas, aunque las pruebas de la interfaz puede ser una fase separada.
- Testeos de aceptación de usuario – esta fase confirma que las necesidades de los usuarios se han cumplido
Es aceptable tener cada vez mayor formalidad aplicada a fases de prueba en términos del grado de documentación de la prueba y las pruebas recogidas.
Informe de validación
El informe de validación resume las actividades de validación asociadas con el desarrollo del sistema. Contiene un resumen de las conclusiones de testeos e informes sobre otros requisitos de validación, por ejemplo, entrenamiento de usuarios y procedimientos actualizados. Proporciona recomendaciones para la aceptación del sistema. El Informe también presenta una estrategia para mantener el sistema en un estado validado. Cuando hay alguna excepción a los entregables requeridos por el plan de validación, éstas se detallan en el informe junto con una justificación. Cualquier acción a ser completada después de la implementación del sistema debe ser rastreada hasta su finalización.
El Informe de Validación debe ser aprobado antes de que el sistema entre en uso operacional.
Liberación / instalación
La liberación e implementación del sistema se basa en el cumplimiento de los entregables de validación y otros requisitos previos necesarios para la gestión del sistema. Estos pueden incluir: una construcción confirmada desde el sistema de gestión de configuración, los procedimientos de instalación, manuales de referencia técnica, manuales de referencia del sistema, manuales de operación del sistema, procedimientos de procesos, capacitación.
En el artículo II, desarrollamos, estrategia de mantenimiento y gestión del cambio, medidas de seguridad, firmas y registros electrónicos, e integridad de datos. para leerlo haga click aquí.
Además para aquellos interesados en el tema, le proponemos un Taller sobre el tema, consúltenos en info@cgmpdoc.com
La validación es mandatoria para los sistemas GxP.
El nivel de la validación dependerá de la configuración del software y de cómo el proveedor diseñó, desarrolló y testeó el sistema computarizado (hardware y software).
La evaluación del proveedor y la categoría del software (de acuerdo a la GAMP5) son claves en el proceso de análisis de riesgo e influenciarán en la determinación de cuanto esfuerzo será requerido para la validación del sistema computarizado.
Les dejo los flujogramas propuestos para las actividades de validación de las categorías 3, 4 y 5, de acuerdo a la GAMP5 (2008).
Flujograma para un sistema de categoría 3.
Flujograma para un sistema de categoría 4 (adaptado).
Flujograma para un sistema de Categoría 5 (a medida).
¿Qué tipos de sistemas computarizados debemos considerar?
En principio los relacionados a las actividades de controles del proceso, los relacionados al laboratorio, las aplicaciones de IT y lo relacionado a infraestructura.
Espero que les haya resultado interesante.
Un sistema computarizado es un software, con su hardware, la gente y los procedimientos.
El resultado de la validación del sistema computarizado, como solemos decir, es la evidencia documentada. Las regulaciones requieren que los sistema computarizados sean validados, a pesar de que definen varios niveles.
La validación de los sistemas computarizados aplica a lo largo del proceso de implementación del proyecto, la operación y el decomiso del mismo.
Disponer de procedimientos y modelos o templates de validación, así como una organización dirigida a validar los sistemas computarizados, son las herramientas claves para estar en cumplimiento.
A este esquema solo quiero agregarles el concepto de ciclo de vida basado en el riesgo:
El enfoque de los ´90 era que independientemente del riesgo que representara cada sistema, los esfuerzos de validación eran idénticos. El ciclo de vida basado en el riesgo, nos lleva a orientar los esfuerzos principalmente en los sistemas que representan algo riesgo y luego los de medio y bajo riesgo.
Esto no representa reducir pasos del modelo en V (Vmodel), SI hacer foco en los sistemas de alto riesgo principalmente.
Espero que les resulte interesante.
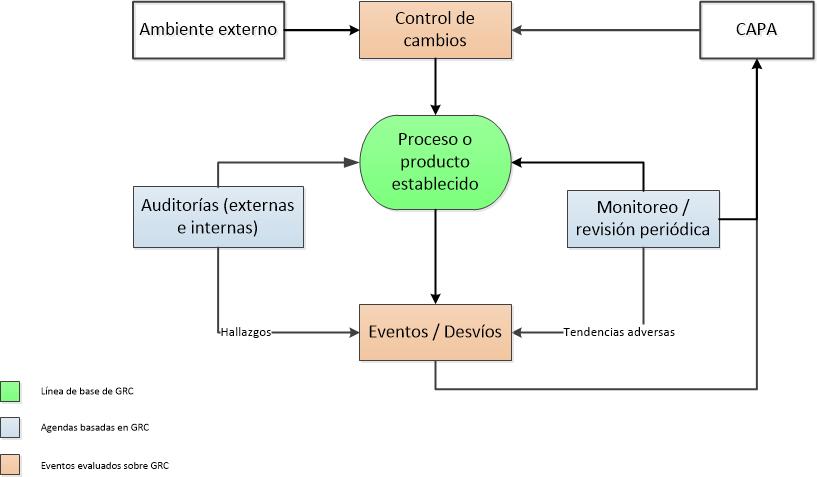
Les dejo algunas preguntas para autoevaluarse respecto de manejo de la GRC:
Ud aplica la GRC para la priorización y la mejora continua y NO para apagar incendios, manejar las crisis o cuando las cosas salen mal?
Ud. Acepta que no existe el riesgo cero?
Todo el personal tiene un claro entendimiento del umbral de riesgo de la empresa?
Los principios y las prácticas de la GRC son entendidas a todos los niveles (desde el Gerente de la Planta hasta los operadores de la misma)?
Ud. usa la GRC a lo largo del ciclo de vida del producto? Por ejemplo:
- Para enfocar sus actividades de validación?
- Para escalar los eventos de calidad?
- Para optimizar sus recursos de auditorías?
- Para clasificar los desvíos o reclamos de clientes?
- Para optimizar el monitoreo ambiental?
- Para proporcionar un marco para la toma de decisiones basado en el riesgo?
Hay un excelente conocimiento de sus procesos y productos a lo largo de su empresa… sin el cual es imposible la GRC?
NUNCA usa la GRC para justificar lo que sabe que es de baja calidad?
Si alguna de las respuestas no fue del todo satisfactoria, Ud. ha detectado una oportunidad de mejora sobre la Gestión de riesgo de calidad, asegúrese tener un plan de acciones de remediación, también desde cGMPdoc podemos proponerle una solución o le dejamos a mano la posibilidad de un taller “In Company” teórico-práctico, consulte en info@cgmpdoc.com
{kind=link}
{kind=link}
Cualquier evaluación de integridad de datos dentro de una operación de laboratorio manual o electrónica debe basarse sobre un análisis de riesgo vs las siguientes características detalladas a continuación:
Atribuible
¿Quién adquirió los datos o realizó una acción y cuándo? Si un registro es modificado por quién y porqué.
Legible
Los datos deben ser registrados de forma permanente (duradera) y fácil de leer.
Contemporáneo
Los datos deben ser registrados al mismo tiempo que el trabajo es efectuado con marca de fecha y hora.
Original
La información registrada deben ser datos originales (datos crudos) o una copia certificada del proceso. Los datos NO deben ser transcritos de una fuente a otra sin justificación y sin control certificado del proceso.
Exacto
Sin errores o ediciones efectuadas, sin enmiendas en los documentos. La información registrada es correcta.
Basado sobre estas características (atributos de integridad de datos) los laboratorios deben asegurar que los procesos analíticos están definidos, mapeados y con los riesgos evaluados para cumplir con la integridad de datos a lo largo del ciclo de vida de los datos. Todos los datos del sistema procesado deben ser incluidos.
El uso de análisis de riesgo y la generación de proceso de mapeo permite una mayor visibilidad de donde la integridad de datos es crítico y además da soporte a las autoinspecciones de tales sistemas.
Dentro de estos requerimientos básicos, las siguientes 10 áreas claves son esenciales para asegurar la integridad de los datos dentro de los límites de un sistema de datos electrónicos, esto está detallado en el siguiente artículo del blog: http://wp.me/p1Hn5Y-tA
Análisis de riesgo:
El análisis de riesgo es el proceso de identificar los peligros y los modos de falla y evaluar las consecuencias potenciales de esos peligros. Esto es críticamente dependiente de que sea involucrado personal con conocimiento.
Los análisis de riesgos de calidad comienzan con una descripción bien definida del problema, una pregunta de riesgo o un análisis de un área de riesgo particular. En el caso de integridad de datos el proceso de laboratorio individual debe ser mapeado en detalle, comenzando por la preparación de la muestra, a través de los resultados de verificación / aprobación y finalizando con el archivo y recuperación de los datos.
Una vez que el proceso fue analizado para las áreas de riesgo crítico de integridad de datos, por ej. Cuál es el riesgo y que impacto podría tener sobre la calidad del producto y la seguridad del paciente, entonces pueden ser asignadas etapas de mitigación.
El proceso de análisis debe seguir las siguientes preguntas, por ej:
- ¿Qué podría salir mal?
- Datos han sido perdidos
- Los sistemas fallan y no hay un plan de continuidad del negocio in place
- Los datos no están siendo registrados
- Los datos no están siendo verificados
- El audit trail no está siendo revisado
- El audit trail no está funcionando
- El entrenamiento y la concientización del equipo/instrumento es inadecuado
- Los passwords están siendo compartidos
- Los resultados no son atribuibles
- ¿Cuál es la probabilidad de que esto salga mal?
- ¿Cuáles son las consecuencias (severidad) para la calidad del producto o la seguridad del paciente?
- La falla ¿Será detectada? ¿Cómo?
Hay muchas herramientas y técnicas que pueden ser usadas para ayudar a identificar peligros y /o modos de fallas y evaluar los riesgos. No hay una sola herramienta o técnica que cumpla con todos los requerimientos, en otro momento ampliaremos sobre las mismas.
Espero que les resulte útil.
Les dejo 13 preguntas para autoevaluarse respecto de manejo de desvíos y sistema CAPA:
- Tiene una cultura abierta, libre de culpables, donde las personas son alentadas a reportar los desvíos?
- Los incidentes son reportados inmediatamente, sin demoras?
- Efectúa una clasificación de los incidentes objetivamente dentro de 24 hs. Para priorizar los recursos y efectuar las investigaciones?
- Inicia sus investigaciones dentro de un día de trabajo hábil, como máximo?
- Investiga proporcionalmente al riesgo, en vez de tratar cada incidente de la misma forma?
- Las investigaciones son efectuadas donde ocurre el incidente, o son manejadas detrás de un escritorio? Siempre?
- Las investigaciones son hechas por personas con “know how” de los procesos y productos, o sólo por QA?
- Los CAPAs están focalizados en prevenir la re-ocurrencia en vez de ocuparse de los síntomas superficiales?
- Implementa CAPAs tan pronto como es posible, en vez de aplicar la “infundada regla de los 30 días”?
- Cree que el error humano es la “consecuencia” y rara vez la “causa” de los desvíos?
- Lleva una tendencia de los eventos repetidos?
- Comparte incidentes y CAPAs a lo largo de la organización para fomentar la mejora continua?
- Arma una agenda para verificar la efectividad de cada CAPA para evaluar cuan exitosos han sido para prevenir la re-ocurrencia del desvío?
Si alguna de las respuestas no fue del todo satisfactoria, Ud. ha detectado una oportunidad de mejora sobre el manejo de los desvíos y sistema CAPA, asegúrese tener un plan de acciones de remediación, también desde cGMPdoc podemos proponerle una solución o le dejamos a mano la posibilidad de un taller “In Company” teórico-práctico.
Dentro de estos requerimientos básicos, les dejamos 10 tips claves, esenciales para asegurar la integridad de los datos dentro de los límites de un sistema de datos electrónicos:

- Identidad única para cada usuario:
Para sistemas basados en papel, siguiendo las Buenas Prácticas de Documentación la identificación de cada analista puede ser alcanzada a través de que cada persona firme o coloque sus iniciales sobre una impresión o notebook de laboratorio. Con los sistemas electrónicos, este principio debe ser mantenido por medio de la identidad única del usuario.
Las cuentas de usuarios no deben ser compartidas, por ej. Una cuenta de usuario establecida para dos analistas para el acceso a un software de laboratorio de un sistema HPLC.
Las identidades de los usuarios no deben ser reusadas, si una persona deja el laboratorio o la compañía, esa identidad del usuario (ID) debe ser removida del uso y cualquier nuevo usuario debe ser habilitado con un nuevo y único número de cuenta de usuario.
- Implementar controles de passwords adecuados:
Los passwords deben ser efectivos de manera que no puedan ser adivinados por otros, por ej. Su nombre / dirección / fecha de cumpleaños, etc. pero no lo demasiado complejos de manera que el usuario lo tiene que tener escrito para poder recordarlo.
Los passwords NO deben ser compartidos bajo ninguna circunstancia. Cuando las credenciales de registro son compartidas, los individuos específicos no pueden ser identificados a través de la actividad de registro y por consiguiente se pierde la integridad de datos.
- Establecer diferentes tipos de usuarios con diferentes privilegios de acceso:
Los accesos a los sistemas o equipos deben ser limitados solo a individuos autorizados, por ej. Analistas, administradores.
Cada tipo de usuario debe ser asignado con diferentes privilegios de acceso de acuerdo al rol que efectuará en el sistema. Debe haber protocolos de seguridad de datos en los cuales se describe el rol del usuario y sus responsabilidades en términos de privilegios de acceso, cambio, modificación, creación y eliminación de proyectos y datos. Solo personal senior debe tener cuentas que le permiten administración completa del sistema, incluyendo la edición de los métodos y de los proyectos.
El rol del administrador del sistema debe asegurar que cualquier requerimiento de cambio, eliminación, etc. es cuidadosamente registrado, y atribuido a un individuo para asegurar que la trazabilidad es mantenida.
- Establecer y mantener una lista de los usuarios actuales e históricos:
Una lista de los usuarios del sistema actuales y de los previos necesita ser establecida y mantenida, si un miembro del personal ha dejado de serlo, entonces la cuenta debe ser inmediatamente puesta como no disponible.
- Control de cambios del sistema:
Relacionado con la asignación de privilegios de accesos está la capacidad de un usuario de hacer cambios. Los cambios deben ser solo efectuados por medio de los individuos autorizados, ej. Cambios de métodos, parámetros de integración y línea de base. Esto debe ser posible para identificar cuáles individuos hicieron los cambios para determinar si ellos tienen el apropiado entrenamiento, educación y experiencia.
- Solamente personal entrenado debe operar el sistema:
Hay un requerimiento para que todo el personal tenga la combinación de educación, entrenamiento y experiencia para efectuar su trabajo. Una parte clave de cualquier programa de entrenamiento es que debe hacer foco sobre la generación de datos y que los cambios solo pueden ser hechos de acuerdo a procedimientos predefinidos para prevenir una acusación de falsificación o fraude.
Los usuarios de instrumentos o equipos deben poder demostrar su capacidad de uso frente a las agencias regulatorias cuando es requerido, dentro de su descripción de rol.
- Registrar toda la información
Los registros de laboratorio deben incluir datos completos derivados de todos los ensayos necesario para asegurar el cumplimiento con las especificaciones y estándares establecidos. Esta es la clave para establecer la integridad de los datos en un sistema de captura de datos electrónicos, a pesar de las situaciones que nos pueden ocurrir, errores, mal funcionamiento de un equipo, etc.
Los datos crudos, “raw data”, (ej. Cromatogramas, pesadas del estándar y de muestras, cálculos, estándares, reactivos, información del instrumento) deben estar disponibles siguiendo la ejecución del trabajo para permitir que una investigación pueda ser llevada a cabo eficientemente y para confirmar la autenticidad y confiabilidad de los datos durante la inspección efectuada por la agencia regulatoria. Todos los datos deben ser registrados de manera que las actividades puedan ser cuidadosamente reconstruidas.
- Definir y documentar registros electrónicos para el sistema
Para registros electrónicos los usuarios deben determinar cuáles datos serán definidos como “raw data”, como mínimo, todos los datos sobre los cuales se basa la toma de decisiones de calidad deben ser definidos como raw data.
Toda la documentación creada durante el análisis la cual incluye un registro es considerada como evidencia de una actividad, de este modo, los archivos de datos creados durante una corrida analítica son registros.
Muchos documentos (instrucciones y/o registros) pueden existir en formatos híbridos, por ej. Algunos elementos como electrónicos y otros en papel. No importa si un registro es generado en papel, existe con firmas manuscritas y le sigue una impresión de un registro electrónico (sistema híbrido) o si es mantenido completamente electrónico.
Controles apropiados deben están en uso para asegurar la integridad de los registros a lo largo del período de retención. Debe estar claramente definido cual registro está relacionado con cada actividad de ensayo y donde es localizado el registro. Controles de seguridad deben estar en uso para asegurar la integridad de los registros a lo largo del período de retención y validado cuando sea apropiado.
- Revisión de las entradas al audit trail para cada lote
Parte de los datos completos de un sistema de datos cromatográfico en las corridas analíticas incluyen el audit trail, y hay un requerimiento bajo las cGMP para la firma o iniciales de una segunda persona para mostrar que el trabajo ha sido hecho correctamente y conforme a los estándares.
Los cambios no deben tapar los datos registrados previamente (sobre escritura).
- Copia de seguridad del Sistema regular
Copias de seguridad (BackUp) regulares de toda la información relevante debe ser llevada a cabo. Típicamente esto se hace diariamente. La integridad y la exactitud de los datos de back up y la capacidad para recuperarlos datos debe ser verificada durante la validación y monitoreada periódicamente. Cuando el back up es efectuado, los registros del back up deben ser chequeados para ver cuando fue efectivo. El back up necesita ser validado, y periódicamente debe efectuarse una recuperación de los datos para ver que el hardware, por ej. CDs, son aún leíbles.