La forma en que Usted ve la situación, cómo la analiza mentalmente y las conclusiones a las que llega, determina el modo en que reacciona al cambio organizativo. Sus pensamientos pueden impulsarle a resistir o hacer que Usted acepte y apoye las nuevas medidas.

Como es natural, los factores que configuran su pensamiento son diversos. Todas las personas interpretan los hechos basándose en la información disponible y en su experiencia, deseos, necesidades, temores, esperanzas, prejuicios y creencias. Filtrar los datos, percibir selectivamente, eligiendo aquello a lo que se prestará atención o que se ignorará es propio de la naturaleza humana. Usted pondera la información de una manera muy personal, evaluando la situación y extrayendo conclusiones que reflejan su punto de vista.
La gente desarrolla con frecuencia una mentalidad negativa basándose en una mala interpretación de las cosas, o en supuestos incorrectos, o en motivos inconfesables o en una forma de pensar equivocada en general. El cambio impacta. La organización se conmueve y cambia. Comienza la ruptura. Los empleados interpretan esta agitación de una manera tergiversada y sacan conclusiones erróneas.
El resultado es una colección de “mitos” respecto del cambio. Estos mitos siempre penetran en la conciencia de la gente y son fuente de problemas. Si Usted está pensando de una manera distorsionada, o no comprende las cosas correctamente, probablemente reaccionará de una forma inadecuada. No abordará el cambio eficazmente.
¿El resultado? Problemas innecesarios.
Es muy importante desafiar los mitos. Primero, deben ser identificados. Luego, deben ser examinados a fondo. Deben ser ponderados a partir de una visión de la realidad más objetiva, mejor informada y más amplia. Los mitos son demasiado dañinos para ignorarlos. Desmotivan a las personas y paralizan la organización.
Les propongo pensar sobre cuáles son los mitos más comunes de la gente en épocas de cambios trascendentales y turbulencia organizativa, envíennos sus comentarios aquí o a info@cgmpdoc.com, nosotros le daremos nuestro punto de vista.
Les dejo uno de los mitos a modo de ayuda:
“puedo seguir haciendo mi trabajo como hasta ahora.”
Pritchett y Pound.
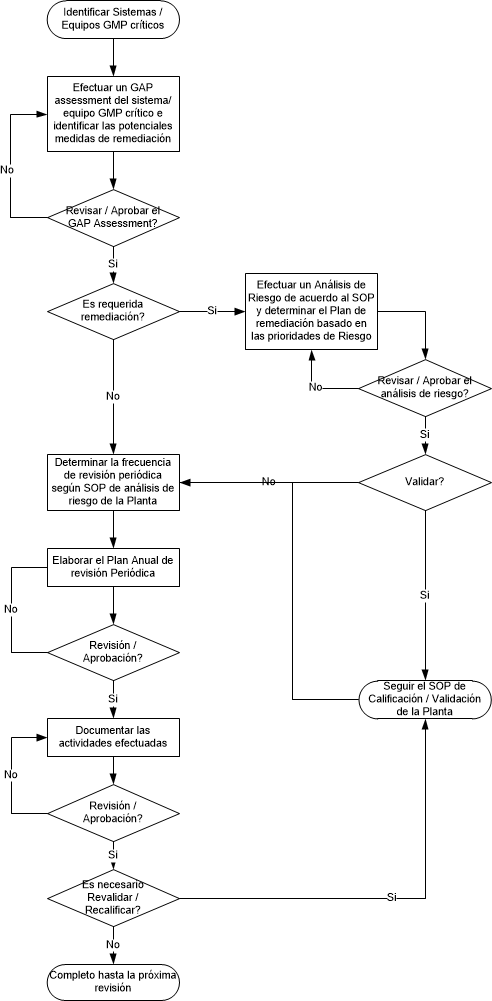
El proceso de auditoria de integridad de datos al laboratorio, requiere de manejar algunos principios, además de un manejo del proceso.
A modo de ejemplo les dejo este esquema de un proceso de HPLC, para que puedan considerar los elementos a evaluar desde el punto de vista de integridad de datos, y además a continuación del mismo, algunos temas a considerar para efectuar la auditoría de integridad de datos, espero que les resulte útil.
NOTA: no considere la preparación de muestra y standars.
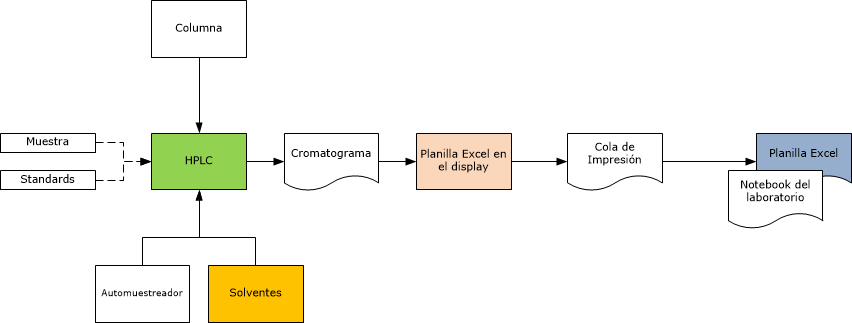
Auditoría de Integridad de datos
- Podríamos decir que cada paso en cualquier proceso de laboratorio tiene un potencial problema de integridad de datos, después de todo los datos son un producto del laboratorio, es como una fábrica de datos
- La herramienta clave del auditor cuando audita la integridad de datos, es el Audit Trail.
- Si hay un Audit trail electrónico disponible, debe ser seguro de auditoría electrónica está disponible debe ser seguro (no susceptible de ser cambiado), accesible, imprimible e inequívocamente asociada con una muestra de análisis en particular.
- Si no hay un Audit trail electrónico disponible, entonces como alternativa se debe trazar todas las actividades a través de las etapas, buscando vínculos entre las etapas y verificando la existencia de todos los documentos o archivos de soporte.
- Otra herramienta es la secuencia de números de análisis, automáticamente asignada a cada inyección (por ej.) en un HPLC. No debe haber archivos perdidos, aunque algunos de ellos serán sólo para propósitos de set-up / preparación.
- En la práctica, hay que seleccionar la más importante para el rastreo de datos para poder efectuar una investigación detallada y siempre incluir cualquier transcripción manual de datos, por ej: ¿Fue correcta? ¿Fue revisada? ¿Cuándo y por quién? ¿Dónde está la evidencia?, etc.
- Desde el punto de vista electrónico, ¿Las planillas de cálculo están controladas? ¿Cómo? ¿Cómo son manejados los cambios de las planillas de cálculo? ¿Fue usada la versión correcta? ¿Está validada? ¿Dónde está el informe? ¿Dónde está la impresión de los resultados (si se hace)?, etc.
Para todos aquellos interesados, envíennos sus respuestas, comentarios o dudas, a info@cgmpdoc.com y con gusto les compartiremos nuestra solución al tema.
Algunas personas se aferran desesperadamente al pasado. Se atan a lo que es familiar y se refugian aún más profundamente en sus cómodas rutinas para no pensar en la espeluznante idea de que tendrán que cambiar. Alguien dijo, “Ninguna organización está fastidiada como para que a alguien no le guste cómo es”. El cambio siempre significa tener que dejar algo, y cuanto mayor sea el sacrificio personal, tanto mayor será el esfuerzo necesario.
Otro motivo que explica porque la gente defiende los viejos hábitos es para mantener la estabilidad personal y sentir que se conserva el control de las cosas. Combaten el cambio por temor al futuro, no por su amor al pasado. Si la incertidumbre y la ambigüedad le provocan ansiedad, será más difícil que el “progreso” despierte su entusiasmo. Cuanto menos le guste lo imprevisible, mayor es la posibilidad que Ud. Proteja el status quo.
Un tercer grupo de gente se resiste al cambio como una forma de desquitarse. Juegan a “penalizar a la empresa” en represalia por los cambios que nos les gustan. Nos referimos aquí a la típica revancha de siempre. Y lo fascinante es observar cómo la gente está dispuesta a perjudicarse a sí misma simplemente por vengarse de la organización.
Finalmente, algunos de los que se resisten al cambio son bien intencionados, son personas que piensan que la empresa está a punto de cometer un error y tienen el valor de intentar corregirlo. Combaten el cambio porque:
- Están totalmente comprometidos en interés de la empresa y
- Tienen la capacidad suficiente para adoptar una postura determinada.
Pero, francamente, estas personas con buenas intenciones a menudo están equivocada. Al tratar de salvar a la empresa en realidad lo que hacen es perjudicarla.
Cuando una empresa inicia cambios, lo hace con una finalidad, no hay ninguna duda de que existen razones urgentes para ello. Casi siempre encontrará una explicación financiera, regulatorias, etc. para lo que está ocurriendo. Estudie la situación:
- ¿Hay acontecimientos externos forzando el cambio en la empresa?
- ¿Debe tomar la organización algunas medidas desagradables para permanecer viva?
- ¿Contribuirá un ejercicio tan duro como éste a desarrollar el músculo de la organización necesario?
Cuando los vientos del cambio golpean su empresa, he aquí un consejo para unos buenos resultados: resistir hace más daño que bien. Para empezar, si adopta una postura opositora, puede tener problemas: alguien puede acusarle de causar problemas, de entorpecer el camino hacia el progreso. Esto puede dañar su carrera.
En segundo lugar, resistirse al cambio requiere esfuerzo, y Ud. Puede hallar formas más productivas de gastar las energías.
Además, probablemente va a perder la batalla de todas maneras. Es más, aunque gane una escaramuza de vez en cuando, perderá la guerra.
En lugar de aferrarse al pasado, agárrese fuertemente al futuro.
Escrito por Price Pritchett y Ron Pound.
Les dejo 5 preguntas para autoevaluarse respecto de integridad de datos:

- Todo su personal entiende que una pobre integridad de los datos, genera pérdida de confianza regulatoria?
- Tiene back up y archivo de sus datos?
- Rutinariamente revisa y comprueba la integridad de los datos?
- Rutinariamente desafía todas las transacciones (audit trail)?
- Tiene validados sus sistemas de manejo de datos?
Si alguna de las respuestas no fue del todo satisfactoria, Ud. ha detectado una oportunidad de mejora sobre la integridad de datos, asegúrese tener un plan de acciones de remediación, también desde cGMPdoc podemos proponerle una solución o le dejamos a mano la posibilidad de un taller “In Company” teórico-práctico de 4 hs de duración.
Estas siete preguntas están dirigidas a verificar si su sistema de control de cambios, gestiona los riesgos, prioriza recursos y esta orientado hacia la mejora continua.

- Su sistema de control de cambios ha sido diseñado con la participación activa de todas las partes interesadas y no solo QA?
- Su sistema de control de cambios es simple y entendible para todos?
- El control de los cambios es considerado vital para la mejora del negocio y la gestión de los riesgos o solo se lo considera por un tema de cumplimiento de las regulaciones?
- El sistema tiene un requerimiento del cambio el cual es revisado y aprobado en hs o días, no meses?
- Su sistema de control de cambios utiliza una evaluación de impacto basada en los riesgos como soporte para la aprobación o el rechazo de las solicitudes de cambios?
- Su sistema rechaza entre el 30-40 % de los requerimientos de cambios para ayudarlo a focalizarse en las prioridades?
- Efectúa el seguimiento de todos los cambios aprobados para asegurarse que los mismos fueron efectivos?
Si alguna de las respuestas no fue del todo satisfactoria, Ud. ha detectado una oportunidad de mejora sobre el sistema de control de cambios, asegúrese tener un plan de acciones de remediación, también desde cGMPdoc podemos proponerle una solución o les proponemos un taller “In Company” sobre el tema, con la teoría y con ejercicios sobre resoluciones de casos de cambios, consúltenos en info@cgmpdoc.com
El objetivo de este artículo es describir un enfoque científico basado en el riesgo para el manejo de la contaminación cruzada, identificar el nivel de riesgo de contaminación cruzada del producto y determinar las acciones de mitigación apropiadas para la protección de:
- La calidad del producto
- El personal
Para:
- La construcción de nuevas instalaciones
- La modificación de las existentes
- La tercerización de la elaboración de productos
Esto es alcanzado en la práctica por medio de la evaluación y documentación de los riesgos de contaminación cruzada y la toma de acciones de mitigación adecuadas, a lo largo del ciclo de vida del producto entero (La elaboración del producto terminado y su packaging), de manera de asegurar que los productos son adecuadamente protegidos de la contaminación cruzada.
Hay aspectos GMP que deben ser cumplidos, y además hay aspectos relacionados a la higiene industrial y el control de la exposición del personal de manufactura que deben ser considerados.
El aumento del portfolio de productos requiere el manejo de un amplio rango de sustancias, algunas de las cuales pueden ser altamente peligrosas. Esto ha resaltado la necesidad de hacer foco sobre identificar y entender claramente los peligros y cómo se manejarán los riesgos de contaminación cruzada y el control adecuado del potencial para el paciente y la exposición del empleado.
Como mencionamos antes, algunos de los factores que afectan la elección de una planta (nueva o existente) son:
- Portfolio de productos (peligro y riesgo)
- Procesos requeridos
- Restricciones de las instalaciones
- Capacidades de los equipos (contención, facilidad de limpieza y mantenimiento)
Vamos a intentar describir las actividades de evaluación necesarias para evaluar el caso donde múltiples productos puedan ser manejados en las mismas instalaciones. Esto también aplica a la introducción de nuevos productos o a la transferencia de productos establecidos a otras plantas.
Por supuesto que el diseño y estrategia preferido es el flexible, disponer de instalaciones multiproductos, lo cual debe tomar en cuenta tanto los requerimientos GMP/ regulatorios como los de higiene industrial.
Para la gestión de riesgo seguimos los lineamientos de la guía de ICH Q9.
Un enfoque claro es necesario para evaluar los riesgos tanto en las áreas de GMP como de HI y definir sobre un criterio científico de manera de permitir tomar la decisión apropiada para el manejo del riesgo.
Debemos balancear las necesidades GMP y las de Higiene industrial (HI), ya que es muy importante asegurar que todos los riesgos, para el producto y para el operador son adecuadamente manejados.
Es sumamente importante relacionar los conceptos que vemos de contaminación cruzada con un proyecto de inversión y el Gantt de las actividades de calificación / validación, sobre todo cuando pensamos en que las plantas tienen que cumplir las exigencias de las cGMP.
Una primera evaluación de alto nivel puede ser efectuada de manera temprana en el proyecto en la fase justificación, cuando la información del producto y del proceso es reunida. Luego en la fase de diseño, el rango del producto, los límites de limpieza, diseño, layout de instalaciones y selección de equipos necesitan ser considerados desde el punto de vista de contaminación cruzada. Más adelante en el proyecto, debe ser efectuado con más detalle un análisis de riesgo de calidad y el enfoque final de diseño y control debe ser establecido antes de la validación. La verificación de que los riesgos de contaminación cruzada son aceptables, serán alcanzados en la validación de limpieza y otros ensayos de control introducidos (para mix up o transferencia mecánica o aérea).
De manera paralela al análisis de riesgo de calidad (GMP), SHE debe efectuar un análisis de higiene industrial (HI) documentado.
Es recomendable efectuar una comparación y revisión de los dos diferentes análisis de riesgos (GMP y HI) una vez que son completados los mismos, para asegurar que cualquier conflicto sea evaluado, por ejemplo: podría haber un conflicto GMP / HI en relación a la dirección del flujo de aire, típicamente el flujo de aire podría ser bajo presión positiva desde un área de manufactura para propósitos GMP de manera de prevenir la entrada al área, pero una presión negativa para propósito de HI para mantener el producto contenido. El diseño final y la solución de control deberán ser acordadas sobre una base caso a caso balanceando ambos requerimientos (GMP y HI).
Ambos criterios GMP e HI son extraídos desde datos toxicológicos y clínicos de fuentes comunes. Cada uno debe tener un enfoque basado en la ciencia para la gestión de riesgo y una clara estrategia para evaluar la exposición potencial.
A pesar que los criterios para ambos requerimientos provienen de los mismos datos toxicológicos y/o clínicos, la forma en que los datos son usados para obtener límites aceptables y las estrategias de control adecuados difieren.
Resumen de diferencias para consideraciones GMP y de HI
| cGMP | Higiene Industrial (HI) | |
| ¿Quién o qué está expuesto? | Producto | Trabajador (operador) |
| Ruta de ingreso | Contaminación cruzada de producto (por medio polvo asentado o producto X retenido en o sobre el producto Y)
Ingestión del paciente, IV (por vía de administración) |
Inhalación
Dérmica A través de membranas mucosas Ingestión |
| Mecanismos de exposición primaria | Retención (inadecuada limpieza)
Mix UP (materiales erróneos) Transferencia mecánica (movimiento de residuos de un objeto a otro) Transferencia aérea (polvo en el aire y contacta con productos o equipos) |
Inhalación (polvo asentado puede ser re-suspendido para ser respirado en otro momento)
Absorción por piel (por contacto o por vía heridas) A través de membranas mucosas (trabajador contaminado toca sus membranas mucosas) Ingestión |
| Bases de estándares para análisis de riesgo | Límites de limpieza
Exposición diaria aceptable (ADE = Acceptable Daily Exposure) |
Límite de exposición ocupacional (OEL= Occupational Exposure Limit) |
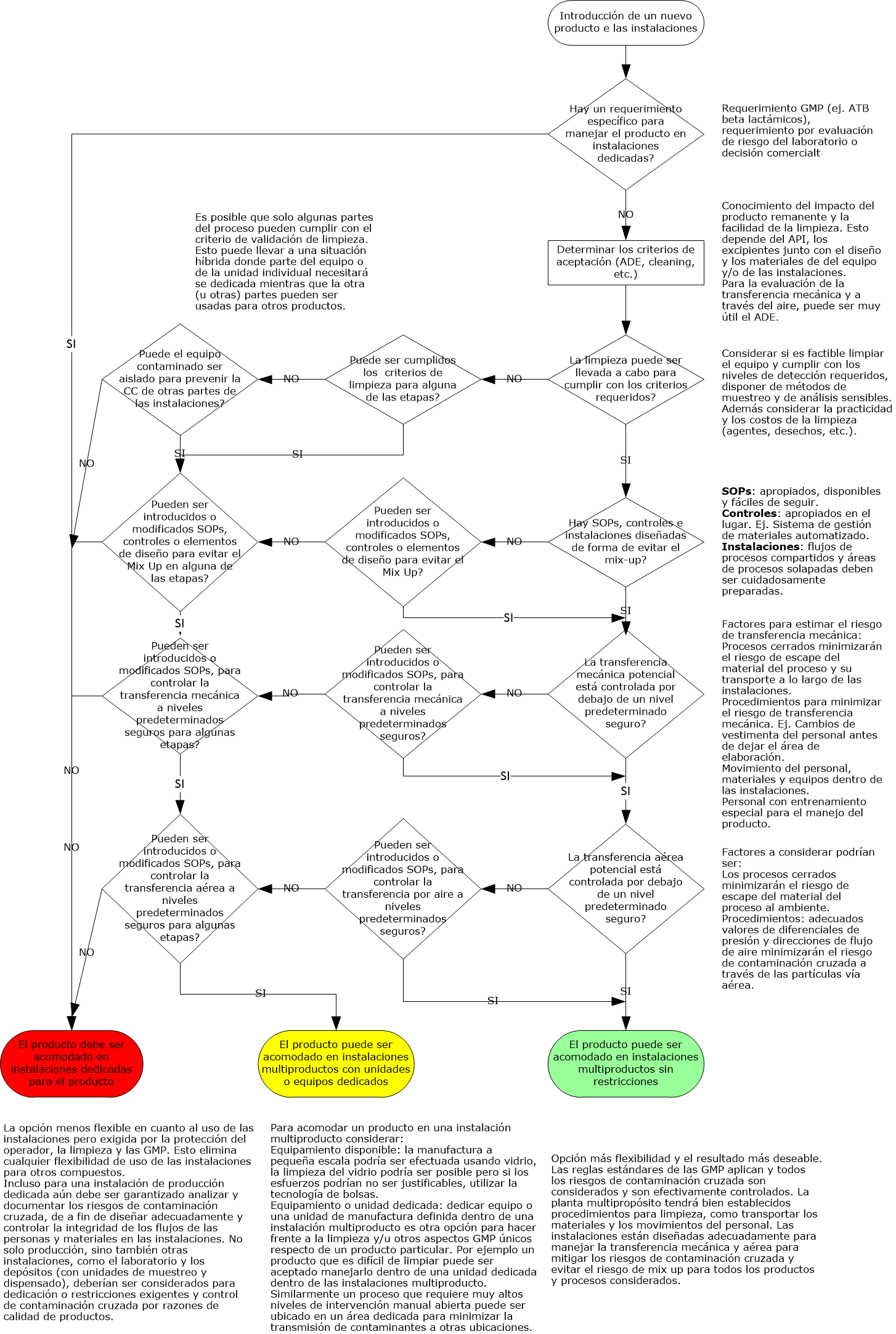
Flujograma GMP de contaminación cruzada GMP: véalo aquí.
Análisis de riesgos GMP
La siguiente información necesitará ser colectada y comprendida para efectuar la evaluación de riesgo:
- Propiedades del producto (ej. Solubilidad, toxicidad, tamaño de partícula) del producto que está en las instalaciones o que será introducido en ellas
- Layout, flujo del proceso, flujo del personal, flujo del aire, diseño de la ventilación
- Diseño del equipamiento y método de operación
- Controles ya existentes en las instalaciones o controles planeados para nuevas instalaciones o productos
- SOPs para control de mix up, limpieza, vestimenta, etc.
Cualquier declaración inicial de alto nivel y decisiones en un proyecto que un cierto producto o tipo de producto no puede ser introducido o empacado en una instalación necesita ser documentado apropiadamente, por ej. En la estrategia de validación y además en las bases del documento de SHE de las instalaciones.
Hay objetivos claves GMP, requerimientos de alto nivel para minimizar el riesgo de contaminación que necesitan ser documentados en URS (requerimientos de usuarios) para asegurar que ellos están incluidos en el proceso de calificación / validación.
Para instalaciones existentes, evaluar que el nuevo producto no será comprometido / contaminado por cualquiera de los productos o procesos existentes en las instalaciones y viceversa, más allá de un límite aceptable.
Para instalaciones nuevas, los productos no serán comprometidos o contaminados en ninguna forma por medio de las actividades de manufactura más allá de un límite aceptable.
Para contaminación cruzada las siguientes cuatro rutas necesitan ser consideradas:
- Mix- Up
- Retención
- Transferencia mecánica
- Aerotransporte
Para ver mas detalle sobre cada una de ellas haga click en Contaminación cruzada.
El impacto sobre el producto / paciente necesita ser evaluado usando la información y conocimientos disponibles.
La competencia en el equipo debería representar especialista con conocimiento en todas estas áreas para que el análisis sea exitoso.
Riesgo: es la combinación de la severidad de las consecuencias potenciales derivada de un peligro (o combinación de peligros) y la probabilidad que esas consecuencias sean realizadas.
Hay diferentes herramientas de análisis de riesgo que pueden ser usadas pero todas ellas deberían incluir factores definidos para evaluar severidad, probabilidad de ocurrencia y detectabilidad y manejar escalas de score comunes.
Severidad: una medida de las posibles consecuencias de un peligro, por ej. Cuán severo podría ser si un paciente / empleado es expuesto a esa peligrosa contaminación? (la escala podría ir desde inadvertido a fatalidad).
Ocurrencia: una medida de la probabilidad que un peligro ocurra. Por ej. Cuán probable es que esa contaminación peligrosa ocurra? (escala desde una ocurrencia en más de 5 años a más de una por lote).
Detectabilidad: la habilidad para descubrir o determinar la existencia, presencia, o hecho de un peligro, por ej. Cuán probable es detectar la contaminación peligrosa antes que la misma sea expuesta al paciente / empleado? (la escala puede ir desde obvio o monitoreado y con alarmas a no detectable).
El propósito del análisis es describir el riesgo, entender la probabilidad de su ocurrencia, si será detectado y evaluar su severidad.
Mitigaciones del riesgo pueden entonces ser determinadas e implementadas. El análisis asistirá en concluir en qué tipo de instalaciones el producto / productos puede ser acomodado. El análisis de riesgo debería ser revisado por medio del proyecto sobre una base regular de manera de asegurar que las mitigaciones son implementadas. El proyecto debería chequear que todas las mitigaciones están “in place” antes de sea entregado para la operación de rutina.
Cuando hay cambios al alcance original del análisis de riesgo el negocio debería revisar el análisis. Si no hay cambios en las instalaciones es recomendado revisar el análisis sobre una base regular, la frecuencia podría tomar en cuenta el portfolio de productos existente.
Espero que le resulte interesante.
Hay 4 rutas potenciales para la contaminación cruzada dentro de las actividades farmacéuticas:
- Mix up: se refiere a la contaminación a un nivel no seguro de un producto con otro. Los altos riesgos de mix up normalmente provienen de fallas cGxP por baja tecnología, debido principalmente a error humano (ej, falla en el seguimiento de los procedimientos) o debilidades de los sistemas (ej. Pobre rotulado de los materiales).
- Retención: es definido como el arrastre de material sobre la superficie en contacto con producto de un producto con otro cuando utilizan el mismo equipamiento en su elaboración. Los factores que afectan la retención están relacionados con la efectividad del procedimiento de limpieza y su aplicación por el personal operativo tras la finalización del proceso, y no solo incluye los residuos de productos, sino también productos secundarios de la limpieza o residuos de materiales de limpieza. El desarrollo de los sistemas de limpieza es una de las áreas de mayor preocupación en la Industria farmacéutica.
- Transferencia mecánica: Esto incluye todas las rutas por medio de las cuales el material puede ser transferido de un producto a otro por contacto, por ej. Ropa o guantes contaminados con un producto que tocan la superficie usada para elaborar un producto diferente, contacto entre la superficie de un producto sucio (contaminado) con otro limpio (accidental).
- Transporte por el aire: Contaminación de un producto a otro por medio de partículas aerotransportadas.
La transferencia asume la generación de un aerosol estable, el cual se mueve a otra área donde se deposita en una cantidad significativa sobre otro producto expuesto (un aerosol es una suspensión de sólidos finos o partículas líquidas en un gas, usualmente aire. ej. Humo). Los sistemas abiertos son los más riesgosos para este tipo de contaminación.

El área de empaque es una de las operaciones de mayor riesgo, en ella suelen ocurrir la mayoría de los problemas que suceden dentro de la manufactura de los productos medicinales.
Los riesgos primarios durante el empaque son:
- Riesgo de mix up
- Riesgo de contaminación cruzada
- Riesgo de contaminación ambiental
- Riesgo de que materiales incorrectos sean llevados a la línea de empaque
- Riesgo de colocar datos variables incorrectos en etiquetas o estuches
- Riesgo de que los blisters y/ o estuches no estén correctamente sellados
El auditor siempre debe tener estos riesgos en mente cuando audita el área de empaque.