Ingresar a Minitab y cargar 20 datos y luego seleccionar la carta CUSUM
Seleccionar las opciones de CUSUM
Seleccionar parámetros, si “s” es conocida por datos históricos, puede ser ingresada, sino Minitab los calcula a partir de los datos ingresados.
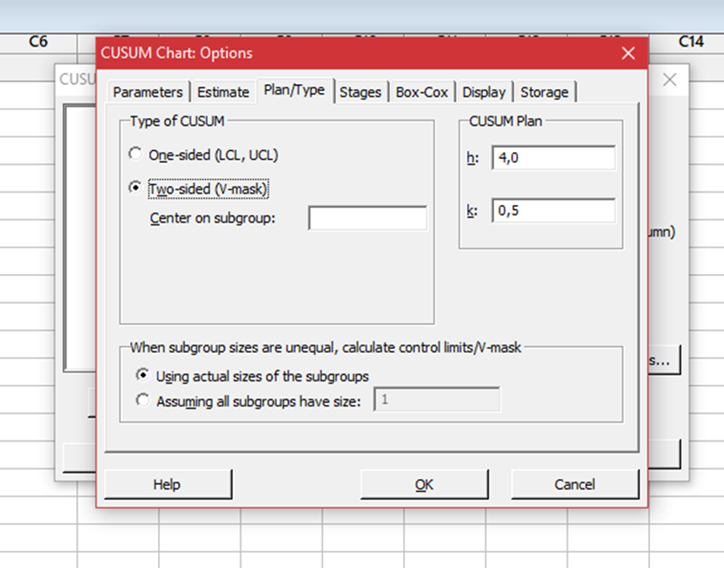
Seleccionar solapa Plan/Tipo y elegir tipo de CUSUM de 2 lados (V-mascara)
Nota
h: Es un valor mayor que 0. Para CUSUM unilaterales, h es el número de desviaciones estándar entre la línea central y los límites de control. Para CUSUM de dos lados (máscara en V), Minitab calcula la mitad del ancho de la máscara en V (H) en el punto de origen por H = h * s. (Puede seleccionar h usando una tabla ARL J.M. Lucas (1976). “The Design and Use of V-Mask Control Schemes,” Journal of Quality Technology, 8, 1-12 o J.M. Lucas and R.B. Crosier (1982). “Fast Initial Response for CUSUM Quality-Control Schemes: Give Your CUSUM a Head Start,” Technometrics, 24, 199-205.
k: Es un valor mayor que 0. Para CUSUM unilaterales, k es la “holgura” permitida en el proceso. Para CUSUM de dos lados, k es la pendiente de los brazos de la máscara en V. Puede seleccionar k usando una tabla ARL.
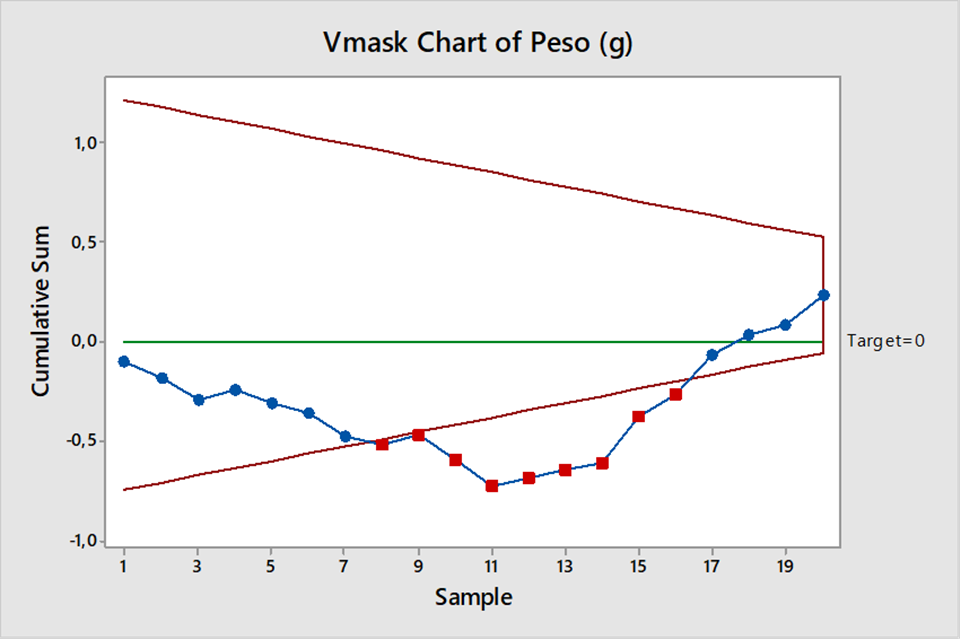
Carta CUSUM donde la suma acumula tanto desviaciones positivas como negativas.
El tipo CUSUM de 2 lados, nos indica que entre el punto 8 y el 16 hay valores fuera de tendencia a investigar, son los que se encuentran fuera de la máscara.
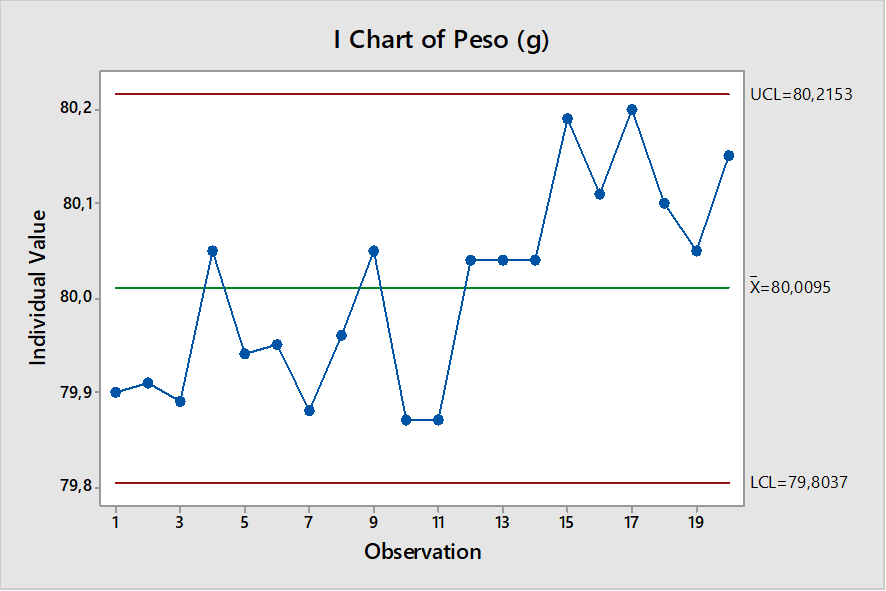
Ahora veamos La carta de control Individual generada a partir de los mismos datos. La misma no muestra puntos fuera de control:
La carta de control Individual no muestra fuera de tendencias. El gráfico CUSUM, muestra valores fuera de control. Concluimos que CUSUM es una carta más sensible, útil para seguir pequeñas variaciones en parámetros críticos.
Luego se debe investigar los puntos fuera de control.
En los sistemas de agua farmacéuticos, el valor TOC (carbono orgánico total) generalmente se determina en línea. Dado que las bacterias también consisten en gran medida en hidrocarburos, surge la pregunta de si también se puede obtener información sobre el estado microbiano del sistema de agua mediante el control de TOC.
Desafortunadamente, esto no es posible. Existen procedimientos rápidos de análisis microbiológicos (sin placas de agar), también para sistemas de agua farmacéuticos. Sin embargo, estos suelen funcionar con otras tecnologías, a menudo mediante medición de partículas y fluorescencia. Sin embargo, no es posible convertir los valores de TOC en recuentos bacterianos. Tampoco es posible sacar conclusiones sobre un posible biofilm en el sistema.
Las bacterias en los sistemas de agua normalmente solo están presentes en una medida mínima en el medio de agua de flujo libre (acuático). Más bien se acumulan en un biofilm que crece en superficies en lugares del sistema con menos flujo (estacionario). Para activar un valor medido en el sistema TOC, los gérmenes tendrían que fluir a través del TOC. Siempre es posible que una parte de un biofilm se desprenda y se mueva a través del sistema por un corto tiempo. Pero incluso entonces es estadísticamente bastante improbable que fluya directamente y de manera reproducible a través del sistema de medición en el momento de la medición. Por lo tanto, los picos a corto plazo en el control de TOC podrían ser causados por un biofilm, pero no tienen por qué serlo. Y para acercarse al valor límite de 500 ppb de TOC, el agua tendría que estar tan contaminada que probablemente ya sería reconocible por la turbidez y el olor.
Por otro lado, un valor muy bajo de TOC en el sistema de agua es un muy buen punto de partida para prevenir la formación de biofilm, ya que la materia orgánica es el alimento de las bacterias que forman del biofilm.
Es común que cuando estamos efectuando actividades de Análisis de Riesgos de Calidad, puedan surgir problemas de subjetividad e incertidumbre.
La mayoría de las herramientas de gestión de riesgos existentes no proporcionan estrategias formales para abordar este tipo de problemas.
Las influencias de lo que se conoce como heurística humana (*) durante las actividades relacionadas con la Gestión de Riesgos de Calidad o GRC (tales como Brainstorming y la estimación de probabilidad de ocurrencia) se pueden añadir a estos problemas.
Los efectos adversos potenciales de tales heurísticas a la hora de identificar modos de falla y sus probabilidades de ocurrencia deben ser contrarrestados.
Una de las fuentes más significativas de incertidumbre y subjetividad en las actividades de GRC es el factor de probabilidad de ocurrencia que suele utilizarse para estimar los riesgos. Muchas definiciones de riesgo incluyen el factor de probabilidad de ocurrencia de la falla.
Esta probabilidad de ocurrencia puede ser estimada de dos formas distintas, algunos lo hacen de manera subjetiva o basada en la creencia que tiene la persona que la falla ocurrirá según su conocimiento actual, o sea que depende del conocimiento de la persona que asigna el valor de probabilidad.
Otros utilizan la frecuencia de ocurrencia de la falla en el pasado (historial de la falla), aquí cuanta más experiencia acumulada hay, mejor. En este punto una limitante es si cuando se está efectuando el análisis, está definida una población relevante de datos de fallas similares y también considerar la disponibilidad de dicho análisis, usualmente colocamos lo que tenemos en mente, dependiendo de nuestra memoria.
Por lo tanto, las actividades de Brainstorming bien diseñadas y basadas en la ciencia presentan oportunidades para reducir la incertidumbre que puede surgir durante la etapa donde se solicita a los expertos que brinden una opinión sobre la probabilidad de que ocurra una falla.
Algunos tipos de heurísticas a considerar son:
Heurísticas de afecto
La teoría de la heurística del afecto es que la respuesta emocional de uno a un estímulo puede afectar las decisiones de un individuo. Cuando las personas tienen poco tiempo para reflexionar y evaluar una situación con cuidado, pueden basar su decisión en sus reacciones emocionales inmediatas. En lugar de realizar un análisis de costo-beneficio, la heurística del afecto se enfoca en provocar una respuesta reaccionaria automática.
Heurística de disponibilidad
Las heurísticas de disponibilidad son juicios que las personas hacen con respecto a la probabilidad de un evento en función de la información que viene a la mente rápidamente. Cuando las personas toman decisiones, generalmente se basan en el conocimiento previo de un evento. Como resultado, tendemos a sobrestimar la probabilidad de que ocurra un evento simplemente porque se nos viene a la mente rápidamente. Tales atajos mentales nos permiten tomar decisiones rápidamente, pero también pueden ser inexactos.
Heurísticas representativas
Las heurísticas representativas ocurren cuando evaluamos la probabilidad de un evento en función de su similitud con otro evento. En general, las personas tienden a sobrestimar la probabilidad de que ocurra un evento en función de su similitud percibida con otro evento. Cuando sucede, tendemos a ignorar la probabilidad real de que ocurra un evento, independientemente de su similitud con otros eventos.
En un próximo artículo veremos algunas ideas para contrarrestar estas heurísticas.
(*) Heurísticas son comportamientos cognitivos que pueden influir en cómo los individuos toman decisiones en un contexto de incertidumbre, y pueden ser una fuente de sesgo significativo o error de juicio.
El entrenamiento es el proceso clave para asegurar que las personas que desarrollan, validan, mantienen, soportan y usan Sistemas Computarizados (SC) tienen la educación, entrenamiento y experiencia para efectuar sus tareas asignadas.
Debe haber procedimientos para entrenamientos que cubran responsabilidades, planes / programas y registros.
El Propietario del proceso de negocios es típicamente el responsable que todos los usuarios estén entrenados.
Personas en posiciones responsables deben tener un entrenamiento apropiado para la gestión y uso del SC dentro del campo de sus responsabilidades. Todos los usuarios y personal de soporte de un sistema GxP, incluyendo el personal contratado, debe ser adecuadamente entrenado, incluyendo entrenamiento GxP básico. Esto además debería tener un entrenamiento específico cubriendo aspectos regulatorios del uso del SC por ej. Aspectos de seguridad o el uso de firmas electrónicas.
Para SC las compañías reguladas deberían entonces:
Establecer necesidades de entrenamientos, incluyendo usuarios, proveedores, data center, departamentos de IT, ingeniería y mantenimiento
Proveer entrenamientos para satisfacer estas necesidades, efectuando la evaluación de la efectividad de los mismos
Asegurar que el personal está consciente de la relevancia e importancia de sus actividades (por ej. GxP)
Asegurar que el personal del proveedor está adecuadamente entrenado, por ej. como parte de la evaluación del proveedor
Mantener los registros de entrenamiento apropiados
Asegurar que el entrenamiento es mantenido actualizado, por ej. Seguimiento de cambios del sistema
Un enfoque basado en el riesgo debe ser usado para mantener el rigor del entrenamiento requerido, medición de la efectividad del mismo, su frecuencia y el enfoque para retención de los registros del entrenamiento.
La nueva revisión ICH Q9 sobre gestión de riesgos de calidad entra en vigencia a partir del 23 de julio de 2023.
¿Cuáles son las novedades de Quality Risk Management (QRM)?
Los objetivos de la revisión fueron mejoras en cuatro áreas:
Subjetividad en las evaluaciones de riesgos y resultados de QRM.
Insuficiente gestión de los riesgos de suministro y disponibilidad de productos
Falta de comprensión de las formalidades de QRM
Falta de claridad sobre la toma de decisiones basada en el riesgo
¿Qué otros cambios importantes se han agregado ahora al documento final? A continuación encontrará un análisis detallado de estos cambios.
La revisión comprende ahora un total de 25 páginas; el documento original tenía 19 páginas. Además de las formalidades sobre gestión de riesgos de calidad (5.1) y toma de decisiones basada en riesgos (5.2) ya mencionadas en el borrador, ahora se ha agregado un subcapítulo sobre gestión y minimización de la subjetividad (5.3). Sin embargo, el nuevo subcapítulo 6.1, el papel de QRM para abordar los riesgos de disponibilidad del producto debido a problemas de calidad/fabricación solo sirve para mejorar la estructura. Hay poco que sea nuevo aquí en términos de contenido. También se mantiene el nuevo punto II.9 Gestión de riesgos de calidad como parte del control/gestión de la cadena de suministro, que ya existía en el borrador.
La introducción ahora es más extensa (1,5 páginas) que en el documento original (1 página). Esto es comprensible, ya que las partes recién agregadas (subjetividad, carencias, etc.) ahora también se abordan en la introducción. El cambio en el proceso de gestión de riesgos de identificación de riesgos a identificación de peligros es nuevo y no ha cambiado desde el borrador.
También es nuevo, en comparación con el documento original una viñeta adicional sobre el tema de la subjetividad en 4.1. Responsabilidades. Sin embargo, esto ya estaba incluido en el borrador del documento. Aparte del borrador del documento y nuevo en comparación con el original, el pasaje sobre subjetividad bajo 4.1 ahora se ha convertido en un punto 5.3 separado.
Gestión de riesgos
En la descripción del proceso de gestión de riesgos, la identificación de riesgos se ha convertido en identificación de peligros en el documento final. Esto también se refleja en el capítulo 7 Definiciones, donde ahora se ha eliminado la identificación de riesgos, como en el borrador del documento. Se hizo un pequeño cambio en la aceptación del riesgo, se eliminó una oración.
En el capítulo 5, Métodos de gestión de riesgos, se agregó una nueva oración en el primer párrafo, que señala que los factores no discretos pueden desempeñar un papel en la detectabilidad. Por lo tanto, los controles de detectabilidad son importantes ya que pueden reducir la probabilidad de ocurrencia.
En el segundo párrafo, se ha eliminado la referencia a las vías informales de análisis de riesgos. La idea detrás de esto parece ser que no se trata de -informal o formal- sino que las transiciones fluidas son posibles en términos de riesgo. Esto también se vuelve a mencionar explícitamente en la introducción del Anexo I, como también podría leerse en el borrador.
También hubo cambios importantes en el Capítulo 5 Métodos de gestión de riesgos a través de la introducción de pasajes sobre la subjetividad. En el documento borrador ya había nuevos subcapítulos sobre formalidades (5.1), sobre decisiones basadas en riesgo (5.2), y ahora, bastante nuevo, en el documento final sobre gestión y minimización de subjetividades (5.3). Como se mencionó brevemente anteriormente, este es un pasaje original de 4.1 del borrador del documento. En general, la toma de decisiones (5.2.) se refiere a la gestión del conocimiento (según ICH Q10) y también a la integridad de los datos en el contexto de las decisiones. En el nuevo capítulo 5.3 se señala la dificultad de evitar la subjetividad y se recomienda considerar siempre este tema cuando se trate de suposiciones o incluso sesgos.
En comparación con el borrador, solo hubo relativamente pocos ajustes textuales en el Capítulo 6 sobre la integración de QRM en la industria y los procesos de autoridad, pero un nuevo subcapítulo, como ya se describió anteriormente. Además, se ha agregado un párrafo sobre la referencia al Anexo II.2, que también puede ser útil en relación con los riesgos para la disponibilidad del producto, en comparación con el borrador del documento y, en consecuencia, también con el documento original.
En general, las referencias (Capítulo 8), especialmente las citas de normas, se han actualizado y se han agregado nuevas referencias bibliográficas.
Cambios en los Anexos
En el Anexo I sobre Métodos y Herramientas QRM, hubo pocos cambios en general. Un error de ortografía en el Capítulo I.5 (HACCP) es digno de mención: haard(s) ciertamente significa peligro(s). En I.9 (Herramientas estadísticas de apoyo), los estándares enumerados se han actualizado con respecto a su problema.
En el Anexo II, el mayor cambio con respecto al documento original es la introducción del Capítulo II.9 (QRM como parte de la gestión/control de la cadena de suministro).
Mientras tanto, la Revisión 1 se ha publicado en el sitio web de la EMA y será válida desde el 23 de julio de 2023.
La semana pasada durante la implementación de un programa de validación de limpieza en un laboratorio, identificamos algunos aspectos sobre los que teníamos que trabajar, y quiero compartir con Uds. uno de los temas, me refiero al uso de detergentes. Está claro que si podemos limpiar nuestros equipos solamente con agua, sin utilizar detergente, es la situación ideal, aunque generalmente esto no sucede.
Les dejo estos TIPS:
La selección del detergente depende del tipo de producto que debe limpiar.
Una vez que valide un detergente, no debería cambiarlo a menos que valide otro.
La validación de un detergente alternativo puede llevar tiempo, dinero y puede afectar el flujo de producción, sin embargo, es una ventaja frente a cualquier contingencia. Además puede ser llevada a cabo de forma gradual.
Establezca con su proveedor un acuerdo de calidad, considerando entre otros temas:
El proveedor debe proporcionarle un método analítico validado para determinar los residuos del detergente.
Comunicación directa y temprana con el fabricante de detergentes para soporte o consultas, notificación de inconvenientes en el abastecimiento del producto, etc.
Cambio de formulación. El proveedor debe notificarlo sobre cualquier cambio de formulación, para que Ud. puede efectuar las evaluaciones del impacto sobre su proceso.
Espero que les resulte útil para revisar su situación actual.
Adaptado del Pharmaceutical on line Newsletter/ 21-feb-2023
Generalmente se utilizan dos técnicas de muestreo en la validación de la limpieza: la prueba de enjuague y la prueba del hisopo. Las siguientes son notas sobre posibles problemas con la prueba de hisopo.
En general, es posible el uso de hisopos para la detección de residuos químicos o microbiológicos. Por lo general, la recuperación de hisopos de microbiología es menor en comparación con las placas de agar para el muestreo de superficie microbiana, en estas notas nos referimos solo al hisopo para la detección de residuos químicos (residuos de productos o detergentes).
Pasos críticos durante la prueba de hisopo:
El propio proceso de muestreo con hisopo, además del análisis, que no se discutirá aquí, tiene dos pasos críticos que deben optimizarse teniendo en cuenta los materiales de la superficie y los residuos:
Paso 1: Recogida de los residuos de la superficie en contacto con el producto utilizando el material del “swab head”.
Paso 2: Transferencia de los residuos del material del “swab head” a la solución de extracción.
Además de una técnica de hisopo reproducible, el material del hisopo en sí mismo es crucial. El uso de hisopos de algodón ya no es “de última generación”. Estos pueden liberar partículas en la superficie de contacto del producto, desintegrarse durante el proceso de hisopado según la técnica de hisopado o no liberar los residuos en la solución de extracción. Los hisopos modernos tienen un material abrasivo que, además de disolver los residuos mediante el uso de una cabeza de hisopo humedecida, también los elimina mecánicamente de la superficie. En este caso, es importante la presión correcta, que se puede comprobar si el hisopo muestra una ligera flexión. En general, son preferibles las superficies de muestreo rectangulares muestreadas en un patrón de control. Para garantizar un área de muestreo correcta, se recomienda pasar el hisopo por carriles superpuestos y muestrear al menos el área de superficie especificada. Más área de superficie hisopada significa un “peor de los casos” y asegura que se genere un resultado bajo falso debido a una superficie más pequeña hisopada. Después de pasar la torunda, debe quedar en la superficie la menor cantidad posible de solución de extracción, que se utiliza para humedecer previamente el cabezal de la torunda. En casos especiales, puede ser útil limpiar la superficie nuevamente con un segundo hisopo seco. Además, debe tenerse en cuenta que después de los procesos de limpieza con medios tibios o calientes o un paso de secado, la superficie del equipo debe enfriarse a temperatura ambiente.
Dependiendo de la técnica de hisopo, la transferencia de los residuos se lleva a cabo secuencialmente en el paso 2. Aquí, el hisopo se agita en la solución de extracción entre los pasos de hisopo individuales. Después del muestreo, el hisopo generalmente se transfiere a la solución de extracción y se deja allí hasta el análisis. Antes de esto, se puede lograr una extracción mejorada adicional utilizando un agitador vibratorio o un baño ultrasónico. La recuperación de muestras se puede mejorar optimizando la solución de extracción. Esto depende principalmente del tipo y condición del residuo. Sin embargo, la solución de extracción en sí también debe ser fácil de limpiar y, además, no interferir con el análisis.
Autor: Dipl.-Ing. (FH) Robert G. Schwarz, publicado en News Letter ECA 1/3/2023
El enfoque del ciclo de vida de los Sistemas Computarizados utilizado por la GAMP5, hace referencia a 4 fases principales:
Concepto
Proyecto
Operación
Retiro
Hoy quiero referirme a la Operación.
Para alcanzar y mantener en cumplimiento un sistema computarizado GxP a lo largo de su ciclo de vida, en primera instancia el mismo debe ser validado (Plan y Reporte de Validación) y luego deben ser aplicados una serie de controles operacionales.
Esto aplica a la mayoría de los sistemas computarizados, cuando el sistema es parte de un proceso de manufactura. Puede ocurrir que no sea requerido una validación del sistema por separado, pero si es necesario verificar la adecuación del sistema computarizado al proceso.
Cuando nos referimos a la operación del sistema computarizado, debemos considerar que no todas las actividades deben ser aplicadas a todos los sistemas. Estas deben ser seleccionadas y escaladas de acuerdo a las características del sistema en cuestión, al riesgo y complejidad del sistema, a través de la aplicación del pensamiento crítico.
Luego de liberar el sistema para su uso, es necesario mantener el estado validado, a través de la gestión de cambios, el mantenimiento del sistema, la actualización de SOPs, mantener la Integridad de datos, efectuar la revisión periódica del sistema a partir del conocimiento adquirido y de la investigación de fallas, el análisis de causa raíz y la generación de CAPAs.
En Julio 2022, fue publicada la segunda edición de la Guía ISPE GAMP® 5: un enfoque basado en el riesgo para sistemas computarizados compatibles con GxP. Esta segunda edición de ISPE GAMP 5 resalta la importancia de los proveedores de servicios, del mayor uso de herramientas de software y automatización para lograr un mayor control, una mayor calidad y menores riesgos a lo largo del ciclo de vida y además destaca la importancia del “pensamiento crítico”. En este punto quiero detenerme y dejarles un resumen de un artículo escrito por Charlie C. Wakeham, sobre Por qué necesitamos el pensamiento crítico: Puede parecer que las únicas cosas que la industria farmacéutica ama más que los acrónimos son las nuevas frases de moda. Y eso puede ser verdad. Pero a veces, detrás de la frase de moda, hay beneficios reales, fuertes y alcanzables para nuestro trabajo, nuestra industria y nuestros pacientes finales. El pensamiento crítico es una de esas frases de moda. Originadas fuera de nuestra industria y adoptadas con entusiasmo por instituciones académicas y consultores a nivel mundial, la mayoría de las definiciones son algo ininteligibles y difíciles de relacionar con la vida real. Charlie menciona que hay no menos de 11 definiciones, pero me voy a quedar con la que para él es su propia definición: Es elegir sabiamente, excluyendo la burocracia, la jerarquía, el ego, el sesgo y la ambición del proceso de toma de decisiones. Se está enfocando en lo que importa, que tiene que ser la calidad en lugar del cumplimiento y la documentación. Obtenga la calidad correcta y el cumplimiento estará allí de todos modos. Me parece más importante lograr que la industria utilice el pensamiento crítico que debatir las complejidades del paradigma. Veamos algunos ejemplos de pensamiento crítico con sistemas computarizados GxP: ESPECIFICACIONES DE REQUISITOS Todos estamos de acuerdo en que necesitamos capturar lo que debe hacer un sistema computarizado cuando estamos planeando un nuevo sistema, es decir, la funcionalidad que proporcionará para respaldar un proceso de negocios en particular. Esto significa que debemos definir qué regulaciones se aplican y qué requisitos individuales deben cumplirse dentro del marco regulatorio general. Necesitamos requisitos para definir la funcionalidad específica que el sistema debe proporcionar en relación con el proceso comercial; cómo controlará una actividad, analizará una muestra, calculará un resultado y almacenará los datos. Puede haber restricciones particulares o requisitos de infraestructura: alojamiento en la nube, funcionamiento en una sala limpia, conexión o interfaz con un sistema existente. Todos estos son tremendamente importantes y deben ser capturados como requisitos. Requisitos sobre cuántos niveles de submenú se permiten en el sistema. Requisitos para “rápido, fácil, fácil de usar”. Estos no tienen sentido para el proceso comercial e inútiles para el paciente final. ¿Qué hay de capturar los requisitos en un documento de Word en lugar de utilizar una herramienta de gestión de requisitos? ¿Qué elección de logotipo, fuente, interlineado? ¿Serán los requisitos firmados a mano o aprobados electrónicamente? Nada de esto tiene ningún impacto en la calidad del producto o la seguridad del paciente. Siempre que los requisitos estén controlados, aprobados, protegidos contra cambios no autorizados, disponibles y actualizados durante toda la vida del sistema, el formato, los medios y la apariencia de la información tampoco tienen impacto en la integridad de los datos. Mencioné “aprobado” en la lista anterior. Revisión por el propietario del proceso y el propietario del sistema, absolutamente. Revisión por otras cuatro personas sin conocimiento de sistemas o procesos que solo querían su nombre en el documento, sin sentido. Mi registro personal en un proyecto CSV era un cliente que demandaba un total de nueve revisores y aprobadores. Si un revisor no va a leer el contenido o entender el contenido, ¿qué representa su firma: un autógrafo para la posteridad? ¿Una confirmación de que creen que el crítico anterior probablemente lo leyó? No ayuda…. PRUEBAS Se pueden obtener grandes beneficios al aplicar el pensamiento crítico a las pruebas y, por lo tanto, no sorprende que la excelente instigación de la FDA de los enfoques de Computer Software Assurance (CSA) tenga un enfoque significativo en las pruebas. Yo estimaría de forma conservadora que los scripts de prueba detallados click a click tardan cinco veces más en escribirse que en ejecutarse. La mayor proporción de defectos encontrados por dichos scripts de prueba son, sin duda, errores en los propios scripts. Veamos la motivación para scripts de prueba tan detallados. Motivación Pensamiento Crítico Debe desafiar una ruta o rama específica en el software; las instrucciones detalladas aseguran que llegue al camino o ramal. Esto aporta valor en la detección de defectos y beneficios finales para la seguridad del paciente. El evaluador no sabe cómo operar el software, por lo que necesita las instrucciones de click a click.
Hoy quiero compartir con Uds, este artículo escrito por Thomas Peither, GMP-Verlag Peither AG
Un sistema CAPA debe entenderse como un elemento importante del sistema de calidad farmacéutica e implementarse de manera uniforme en toda la empresa o grupo. En principio, se puede implementar de dos formas diferentes: como un sistema autónomo o como un sistema integrado. En este artículo, profundizaré en los pros y los contras de cada uno, y también compartiré algunas de las mejores prácticas generales de CAPA.
Sistemas CAPA Autónomos
Un sistema CAPA autónomo se caracteriza por su propia documentación y seguimiento de horarios. Un SOP autónomo describe el proceso para la definición, responsabilidad, seguimiento del cronograma y cierre de las actividades CAPA. Estas se derivan como posibles actividades de seguimiento a incidencias que están reguladas en otros sistemas, como por ejemplo para la atención de desviaciones, queja / reclamos o autoinspecciones.
En base a una queja, por ejemplo, se inicia un proceso CAPA en el sistema CAPA independiente después de su evaluación de riesgos, si es necesario, y se realiza un seguimiento mediante un sistema de documentación separado.
Los sistemas autónomos tienen una ventaja decisiva: se pueden tomar medidas inmediatas para eliminar la desviación y aún se pueden documentar y procesar correcciones rápidas dentro del sistema de activación, evitando así una generación inflacionaria de procesos CAPA.
Otra ventaja de un sistema autónomo es una clara separación entre el sistema CAPA y otros procesos. Por lo tanto, cada uno de los sistemas puede ser monitoreado, documentado y discutido por sí solo.
Sin embargo, esto también conlleva una desventaja: los dos procesos separados deben referenciarse entre sí para vincularlos de manera clara y comprensible. Esto siempre conlleva el riesgo de que la información se pierda o se transfiera incorrectamente.
Además, eventos similares que pueden no ser espectaculares cuando se ven individualmente pueden convertirse en debilidades del sistema cuando se ven como un todo, lo que es más difícil de reconocer cuando se ven por separado.
Los sistemas de IT son, por supuesto, útiles aquí. Cada uno genera procesos específicos para las diferentes actividades, pero están interrelacionados y hacen accesible toda la información del otro proceso.
Sistemas CAPA Integrados
Un sistema CAPA integrado suele ser algo más simple. Esa es una clara ventaja. Aquí, al final del procesamiento del incidente inicial, se definen las medidas CAPA correspondientes en la misma hoja de documentación y posteriormente también se documenta su implementación. Después de manejar tanto el problema como las medidas CAPA, la unidad de calidad responsable puede evaluar y cerrar ambos procesos juntos.
Esta ventaja, por otro lado, hace que el proceso sea más complejo: una vez finalizado todo el proceso, se debe transferir de nuevo a la unidad de calidad para su evaluación estadística (análisis de tendencias, revisión por la dirección, etc.) y cierre en el sistema de gestión de calidad. Si hubiera inquietudes por parte de la unidad de calidad, el sistema debe otorgar a la unidad de calidad un derecho de rechazo. En este caso, el procedimiento será transferido nuevamente a la unidad responsable para lograr una conclusión aceptable.
Aspectos importantes para ambos sistemas CAPA
Autónomo o integrado, para ambas variantes, la descripción del sistema CAPA debe hacerse en el manual de gestión de la calidad junto con la descripción de los sistemas para desviaciones, quejas, retiros, etc.
Además, un sistema CAPA requiere su propio SOP en el que se definen los procedimientos junto con las responsabilidades y la documentación necesaria.
Tiene sentido prescribir un análisis de riesgo posterior a la investigación y procesamiento de las desviaciones y, en consecuencia, la decisión sobre el posible inicio de una actividad CAPA.
No importa a qué resultado se llegue, en todo caso se documenta que se ha pensado en una medida más en el sentido de CAPA.
Tres aspectos son importantes aquí:
Seguimiento rápido de la implementación,
Control de eficacia y
Informar las actividades de CAPA en la revisión por la dirección.
¿Cuáles son las características de un sistema CAPA eficaz?
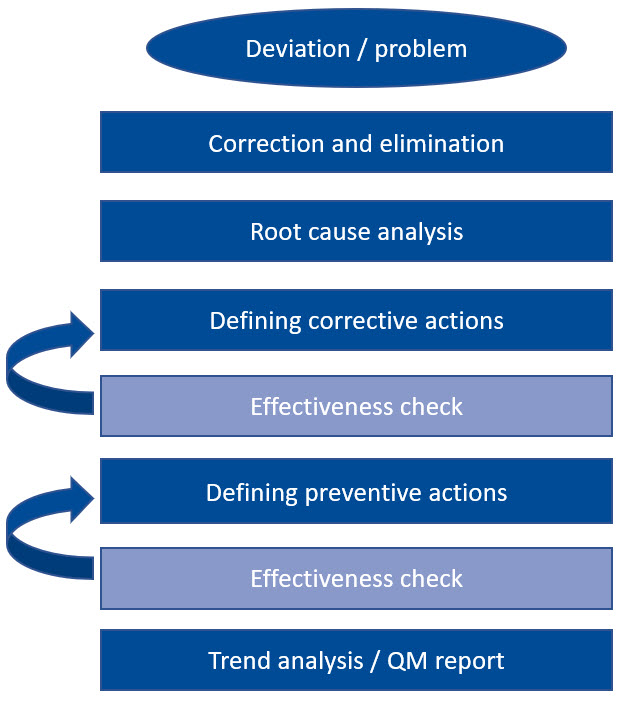
En última instancia, todos los sistemas CAPA efectivos se basan en los mismos procesos. Al definir las medidas CAPA, es importante determinar cómo se puede comprobar su eficacia. Para ello, se necesitan criterios objetivos que puedan utilizarse para determinar el resultado. Si la medida tomada no muestra el éxito esperado, se debe considerar una nueva acción correctiva o preventiva. Por lo tanto, un sistema CAPA es un proceso iterativo continuo que sigue hasta que conduce a un resultado satisfactorio.
Veamos un ejemplo: la primera acción es siempre la corrección y eliminación inmediatas de la desviación, seguida inmediatamente por una investigación de la causa del error. Una vez que se ha identificado la causa, se definen las acciones correctivas apropiadas para evitar que vuelva a ocurrir. Una vez que estas medidas hayan demostrado ser efectivas y sostenibles, este proceso estará completo.
En principio, lo mismo se aplica a las acciones preventivas. Solo cuando se ha comprobado su eficacia se realiza un análisis de tendencias y todo se registra en el informe de gestión de calidad. Un enfoque tan rígido y pragmático no solo suena factible, sino que también es extremadamente práctico.
Hay varias razones por las que los sistemas CAPA, sin embargo, se han descarrilado con bastante facilidad en algún momento o todavía están en marcha. Por ejemplo, como suele ser el caso cuando se introduce un nuevo paradigma en la garantía de calidad farmacéutica, se observó un enorme aumento en las medidas CAPA en el período inicial cuando se establecieron los sistemas CAPA.
Esto se debió sólo en parte a una mayor conciencia de las interrelaciones pertinentes. Mucho más frecuentemente, se debió simplemente a malentendidos, como la evaluación de que toda no conformidad debe necesariamente resultar en una medida CAPA. Sin embargo, el Capítulo 1 de la Guía GMP de la UE no estipula la investigación de desviaciones o el inicio de medidas CAPA para cada no conformidad, sino que las limita a las desviaciones “relevantes”. Por lo tanto, es importante brindar a los empleados la capacitación suficiente y utilizar ejemplos para aumentar su conciencia sobre qué es una desviación y cómo debe documentarse.
Si una desviación documentada en el sitio se procesa más en el sistema CAPA o se puede corregir rápidamente en el sitio, lo decide una unidad superior o el control de calidad. En todo caso, deberá estar regulado en el SOP asociado.
Los involucrados en el proceso definitivamente deben participar en la evaluación de las no conformidades, porque conocen los procesos y las interrelaciones, tienen experiencia con las posibles causas y deben poder tomar decisiones rápidas.
La evaluación o clasificación rápida de un evento en particular no siempre tiene que ser realizada por un departamento de calidad superior. El experto en el sitio está, al menos según el entendimiento europeo de GMP, igualmente autorizado para ayudar a tomar decisiones. Como suele ser el caso, las mejores decisiones las toman todas las partes involucradas.
Es deseable, pero hasta ahora solo realizado en unas pocas empresas, vincular el instrumento de acción preventiva prospectiva con otros métodos de mejora continua de procesos como Six Sigma o Excelencia Operacional. Entonces, en lugar de usarse de forma rutinaria para cada desviación, en realidad solo podría aplicarse específicamente y con alta prioridad después de la evaluación de riesgos.
3 consejos para la vida diaria de GMP
Haga la vida un poco más fácil para los tomadores de decisiones y agregue una selección de medidas CAPA fijas al final del SOP para manejar las desviaciones, como cambios en las instrucciones de trabajo, cambios en los procesos, aumento en las frecuencias de prueba o monitoreo, cambio en las responsabilidades, o reentrenamiento de los empleados.
Use la medida estándar de “reentrenamiento de empleados” de uso frecuente con moderación. Una instrucción breve y directa a los empleados como medida correctiva suele ser más eficaz que una nueva formación formal.
Si la misma desviación o una similar ocurre una y otra vez, es hora de mirar más de cerca. Haga correcciones reales, tómese el tiempo para reescribir un SOP confuso y desordenado. Procesos, procedimientos, pautas y documentación más simples a menudo traen mejoras duraderas.
Este artículo se basa en el conocimiento de GMP contenido en el portal en línea GMP Compliance Adviser, que brinda información detallada sobre las mejores prácticas y regulaciones de GMP de Europa, EE. UU., Japón y muchos más (PIC/S, ICH, OMS, etc.). ).
Sobre el Autor:
Thomas Peither es miembro de la junta y director de marketing y desarrollo empresarial de GMP-Verlag Peither AG, Schopfheim, Alemania. Desde 2008, también dirige GMP Publishing Peither, Inc. Tiene más de 20 años de experiencia como consultor de GMP para empresas farmacéuticas. Se le puede contactar en thomas.peither@gmp-verlag.de.