Hoy quiero compartir con Uds, este artículo escrito por Thomas Peither, GMP-Verlag Peither AG

Un sistema CAPA debe entenderse como un elemento importante del sistema de calidad farmacéutica e implementarse de manera uniforme en toda la empresa o grupo. En principio, se puede implementar de dos formas diferentes: como un sistema autónomo o como un sistema integrado. En este artículo, profundizaré en los pros y los contras de cada uno, y también compartiré algunas de las mejores prácticas generales de CAPA.
- Sistemas CAPA Autónomos
Un sistema CAPA autónomo se caracteriza por su propia documentación y seguimiento de horarios. Un SOP autónomo describe el proceso para la definición, responsabilidad, seguimiento del cronograma y cierre de las actividades CAPA. Estas se derivan como posibles actividades de seguimiento a incidencias que están reguladas en otros sistemas, como por ejemplo para la atención de desviaciones, queja / reclamos o autoinspecciones.
En base a una queja, por ejemplo, se inicia un proceso CAPA en el sistema CAPA independiente después de su evaluación de riesgos, si es necesario, y se realiza un seguimiento mediante un sistema de documentación separado.
Los sistemas autónomos tienen una ventaja decisiva: se pueden tomar medidas inmediatas para eliminar la desviación y aún se pueden documentar y procesar correcciones rápidas dentro del sistema de activación, evitando así una generación inflacionaria de procesos CAPA.
Otra ventaja de un sistema autónomo es una clara separación entre el sistema CAPA y otros procesos. Por lo tanto, cada uno de los sistemas puede ser monitoreado, documentado y discutido por sí solo.
Sin embargo, esto también conlleva una desventaja: los dos procesos separados deben referenciarse entre sí para vincularlos de manera clara y comprensible. Esto siempre conlleva el riesgo de que la información se pierda o se transfiera incorrectamente.
Además, eventos similares que pueden no ser espectaculares cuando se ven individualmente pueden convertirse en debilidades del sistema cuando se ven como un todo, lo que es más difícil de reconocer cuando se ven por separado.
Los sistemas de IT son, por supuesto, útiles aquí. Cada uno genera procesos específicos para las diferentes actividades, pero están interrelacionados y hacen accesible toda la información del otro proceso.
- Sistemas CAPA Integrados
Un sistema CAPA integrado suele ser algo más simple. Esa es una clara ventaja. Aquí, al final del procesamiento del incidente inicial, se definen las medidas CAPA correspondientes en la misma hoja de documentación y posteriormente también se documenta su implementación. Después de manejar tanto el problema como las medidas CAPA, la unidad de calidad responsable puede evaluar y cerrar ambos procesos juntos.
Esta ventaja, por otro lado, hace que el proceso sea más complejo: una vez finalizado todo el proceso, se debe transferir de nuevo a la unidad de calidad para su evaluación estadística (análisis de tendencias, revisión por la dirección, etc.) y cierre en el sistema de gestión de calidad. Si hubiera inquietudes por parte de la unidad de calidad, el sistema debe otorgar a la unidad de calidad un derecho de rechazo. En este caso, el procedimiento será transferido nuevamente a la unidad responsable para lograr una conclusión aceptable.
Aspectos importantes para ambos sistemas CAPA
Autónomo o integrado, para ambas variantes, la descripción del sistema CAPA debe hacerse en el manual de gestión de la calidad junto con la descripción de los sistemas para desviaciones, quejas, retiros, etc.
Además, un sistema CAPA requiere su propio SOP en el que se definen los procedimientos junto con las responsabilidades y la documentación necesaria.
Tiene sentido prescribir un análisis de riesgo posterior a la investigación y procesamiento de las desviaciones y, en consecuencia, la decisión sobre el posible inicio de una actividad CAPA.
No importa a qué resultado se llegue, en todo caso se documenta que se ha pensado en una medida más en el sentido de CAPA.
Tres aspectos son importantes aquí:
- Seguimiento rápido de la implementación,
- Control de eficacia y
- Informar las actividades de CAPA en la revisión por la dirección.
¿Cuáles son las características de un sistema CAPA eficaz?
En última instancia, todos los sistemas CAPA efectivos se basan en los mismos procesos. Al definir las medidas CAPA, es importante determinar cómo se puede comprobar su eficacia. Para ello, se necesitan criterios objetivos que puedan utilizarse para determinar el resultado. Si la medida tomada no muestra el éxito esperado, se debe considerar una nueva acción correctiva o preventiva. Por lo tanto, un sistema CAPA es un proceso iterativo continuo que sigue hasta que conduce a un resultado satisfactorio.
Veamos un ejemplo: la primera acción es siempre la corrección y eliminación inmediatas de la desviación, seguida inmediatamente por una investigación de la causa del error. Una vez que se ha identificado la causa, se definen las acciones correctivas apropiadas para evitar que vuelva a ocurrir. Una vez que estas medidas hayan demostrado ser efectivas y sostenibles, este proceso estará completo.
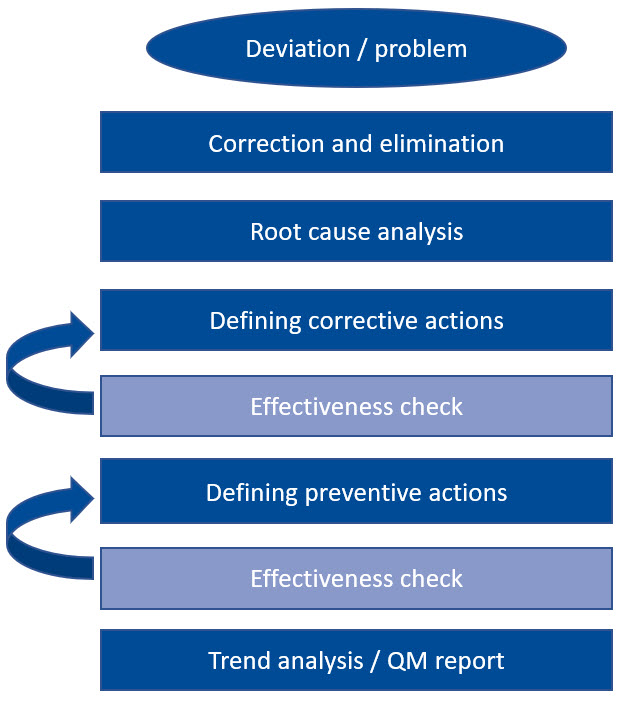
En principio, lo mismo se aplica a las acciones preventivas. Solo cuando se ha comprobado su eficacia se realiza un análisis de tendencias y todo se registra en el informe de gestión de calidad. Un enfoque tan rígido y pragmático no solo suena factible, sino que también es extremadamente práctico.
Hay varias razones por las que los sistemas CAPA, sin embargo, se han descarrilado con bastante facilidad en algún momento o todavía están en marcha. Por ejemplo, como suele ser el caso cuando se introduce un nuevo paradigma en la garantía de calidad farmacéutica, se observó un enorme aumento en las medidas CAPA en el período inicial cuando se establecieron los sistemas CAPA.
Esto se debió sólo en parte a una mayor conciencia de las interrelaciones pertinentes. Mucho más frecuentemente, se debió simplemente a malentendidos, como la evaluación de que toda no conformidad debe necesariamente resultar en una medida CAPA. Sin embargo, el Capítulo 1 de la Guía GMP de la UE no estipula la investigación de desviaciones o el inicio de medidas CAPA para cada no conformidad, sino que las limita a las desviaciones “relevantes”. Por lo tanto, es importante brindar a los empleados la capacitación suficiente y utilizar ejemplos para aumentar su conciencia sobre qué es una desviación y cómo debe documentarse.
Si una desviación documentada en el sitio se procesa más en el sistema CAPA o se puede corregir rápidamente en el sitio, lo decide una unidad superior o el control de calidad. En todo caso, deberá estar regulado en el SOP asociado.
Los involucrados en el proceso definitivamente deben participar en la evaluación de las no conformidades, porque conocen los procesos y las interrelaciones, tienen experiencia con las posibles causas y deben poder tomar decisiones rápidas.
La evaluación o clasificación rápida de un evento en particular no siempre tiene que ser realizada por un departamento de calidad superior. El experto en el sitio está, al menos según el entendimiento europeo de GMP, igualmente autorizado para ayudar a tomar decisiones. Como suele ser el caso, las mejores decisiones las toman todas las partes involucradas.
Es deseable, pero hasta ahora solo realizado en unas pocas empresas, vincular el instrumento de acción preventiva prospectiva con otros métodos de mejora continua de procesos como Six Sigma o Excelencia Operacional. Entonces, en lugar de usarse de forma rutinaria para cada desviación, en realidad solo podría aplicarse específicamente y con alta prioridad después de la evaluación de riesgos.
3 consejos para la vida diaria de GMP
Haga la vida un poco más fácil para los tomadores de decisiones y agregue una selección de medidas CAPA fijas al final del SOP para manejar las desviaciones, como cambios en las instrucciones de trabajo, cambios en los procesos, aumento en las frecuencias de prueba o monitoreo, cambio en las responsabilidades, o reentrenamiento de los empleados.
Use la medida estándar de “reentrenamiento de empleados” de uso frecuente con moderación. Una instrucción breve y directa a los empleados como medida correctiva suele ser más eficaz que una nueva formación formal.
Si la misma desviación o una similar ocurre una y otra vez, es hora de mirar más de cerca. Haga correcciones reales, tómese el tiempo para reescribir un SOP confuso y desordenado. Procesos, procedimientos, pautas y documentación más simples a menudo traen mejoras duraderas.
Este artículo se basa en el conocimiento de GMP contenido en el portal en línea GMP Compliance Adviser, que brinda información detallada sobre las mejores prácticas y regulaciones de GMP de Europa, EE. UU., Japón y muchos más (PIC/S, ICH, OMS, etc.). ).
Sobre el Autor:
Thomas Peither es miembro de la junta y director de marketing y desarrollo empresarial de GMP-Verlag Peither AG, Schopfheim, Alemania. Desde 2008, también dirige GMP Publishing Peither, Inc. Tiene más de 20 años de experiencia como consultor de GMP para empresas farmacéuticas. Se le puede contactar en thomas.peither@gmp-verlag.de.
Deje una Respuesta