¿Cómo conducir una revisión anual de productos efectiva?
Antes de comenzar quiero volver sobre una idea mencionada anteriormente y que supongo la habrán escuchado muchas veces y en distintos lugares: el APR es más que un requerimiento regulatorio, es una revisión que le permite al elaborador entender más su proceso y tomar acciones de mejora.
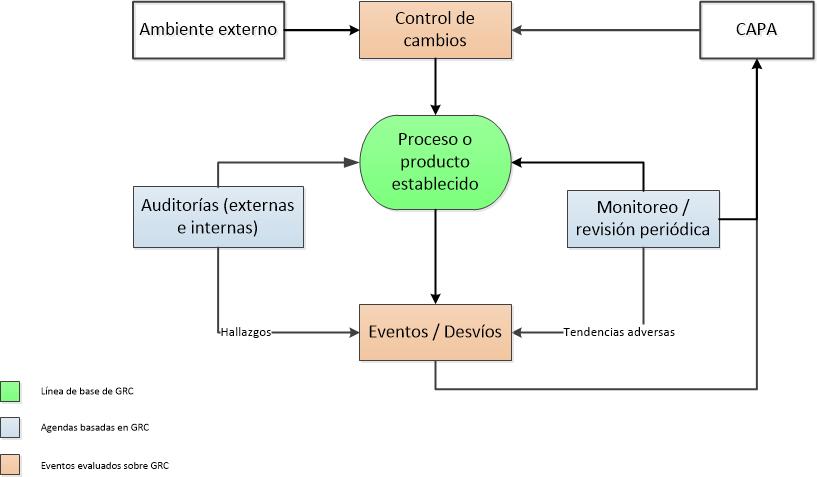
Las cGMP necesitan una evaluación anual de los estándares de calidad de los productos para determinar la necesidad de ajustar sus especificaciones y los procedimientos de elaboración y de control.
Las guidelines acentúan la importancia del análisis de las investigaciones o desvíos conducidos y los reclamos de productos recibidos, mientras el reporte de APR debe explorar, en profundidad, las causas de recalls y de devoluciones de productos, si las hubiera.
De manera general, el mandato de las Agencias regulatorias (por ej. FDA) es mirar a través y sistemáticamente por todas las áreas de mejora y alinear los procesos consistentemente a la elaboración de productos de calidad.
Por estas mismas razones generales, las GMP requieren a los elaboradores analizar previa revisión, examinar los resultados del análisis de productos terminados y los controles en procesos críticos y revisar: lotes fallidos, desvíos, CAPAs, controles de cambio, estudios de estabilidad, devoluciones, reclamos, calificaciones en equipos críticos y acuerdos de calidad.
Los CAPAs de la revisión anual de productos necesitan ser comunicados al Senior Management y completados de manera efectiva y a tiempo, su efectividad debe ser chequeada por medio de las auditorías internas.
Estructura de un informe de APR:
Puede variar según la compañía, o incluso el producto. La idea de seguir un template o modelo asegura que todos los aspectos requeridos son evaluados.
Pero un APR es un documento que evoluciona.
Estructura de informe de APR: http://wp.me/p1Hn5Y-fs
Cambios y correcciones
La Revisión de los cambios puede ser desglosada en: cambios de materia prima, en los componentes de empaque, en especificaciones y documentos maestros. La sección de no conformidades y desvíos necesita ser revisada sobretodo considerando las acciones correctivas y su efectividad. Cualquier CAPA vencido o inefectivo necesita ser discutido en el informe. Este es uno de los aspectos fundamentales.
Cualquier tendencia observada necesita ser direccionada, no solo las que son OOS.
Racionalización de los datos de origen y administración del APR
Compilar los datos crudos es siempre un esfuerzo de equipo, pero el departamento de QA debe tomar el liderazgo y ser el último responsable de la administración del programa.
Un comité de APR podría típicamente incluir un representante de QA, QC, validaciones, operaciones, estabilidad, ingeniería, y logística. Un borrador de informe es completado sobre el análisis crítico de los datos crudos, luego se discute en una reunión del comité de APR para determinar los CAPAs efectivos.
Otro desafío para el administrador de APR es la recuperación de datos para el propósito de la revisión.
Las empresas con sistemas de adquisición de datos calificados pueden usar sus bases de datos, mientras los elaboradores que se manejan con papeles pueden tener que revisar los documentos de lote en forma individual para ver los parámetros del proceso, los controles en proceso, los análisis finales, los rendimientos, etc. En cualquiera de las dos formas, los datos crudos usados para el análisis deben ser exactos de manera de completar una análisis/evaluación efectivo. Si son observadas desviaciones del proceso durante la revisión, puede ser requerido colectar información adicional para justificar dichos hallazgos.
Los datos deben estar disponibles para el administrador del APR en el momento oportuno. Todos ellos deben ser verificados por una segunda persona si son colectados manualmente.
Si son utilizadas planillas de cálculo, las mismas deben estar validadas previo a su uso.
Conclusiones
Efectuar un APR es un requerimiento para los mercados regulados. Pero más que eso, la revisión ayuda al elaborador a entender y conocer mejor sus procesos y para reunir información adicional para posteriores mejoras.
Es una enorme ayuda en la determinación si un producto aún cumple las especificaciones, si necesita un cambio de formulación, modificación del empaque, una revisión de especificación, o un proceso más robusto. Una conclusión de APR es un paso previo al desarrollo futuro del producto y por lo tanto debería ser exacto y respaldado mediante datos adecuados.
Las guías de validación de procesos de la FDA piden la verificación continua del proceso. Así, un programa de APR puede servir como un sistema on going (etapa 3: verificación continua del proceso) para colectar y analizar los datos del producto / proceso que relaciona la calidad del producto.
Las necesidades del APR son parte del plan de mitigación de riesgos de acuerdo a las recomendaciones del ICH Q9.
La información reunida y las tendencias observadas pueden ayudar al desarrollo de un nuevo producto como tal y entonces esto es esencial para distribuir el reporte a todas las partes pertinentes e interesadas. El esfuerzo puede además ser revisado y compartido con equipos de mejora continua (lean process) mientras el desarrollo de CAPAs de un APR son críticos en evitar riesgos potenciales para un producto en el futuro.
Les dejo un artículo adicional:http://wp.me/p1Hn5Y-f3
Desde cGMPdoc, le ofrecemos la posibilidad de una promoción compuesta de 1 SOP para el manejo de los APR el cual incluye un informe modelo y una capacitación powerpoint con notas aclaratorias y su respectivo cuestionario de evaluación, consulte en info@cgmpdoc.com.
Además si Ud. Lo necesita podemos ofrecerle efectuar la revisión anual de sus productos, consultenos aquí.