Continuando con el artículo anterior sobre análisis de tendencias de atributos de calidad.
Las tendencias manuales deben ser efectuadas usando las cartas de comportamiento del proceso. Así podemos evaluar si un proceso está bajo control estadístico o fuera de control estadístico, además de un análisis visual sobre la capacidad del proceso.
Aplicando ciertas reglas a los patrones en los datos de la carta de comportamiento del proceso ayudará a evaluar si el proceso es estable y está en control. Estas reglas están referenciadas como las Western Electric Pattern Rules (WEP rules). Cuando son aplicadas on time, las violaciones a las reglas específicas pueden proveer una percepción del proceso para ayudar a la toma de CAPAs inmediatas.
Las reglas WEP son las siguientes:
- Un punto más allá de la zona A
- Secuencia de 9 puntos en C o más allá (de un mismo lado de la línea central)
- Secuencia de 6 puntos en ascenso o descenso
- Secuencia de 14 puntos alternando arriba y abajo
- Secuencia de 3 puntos con 2 en A
- Secuencia de 5 puntos con 4 en B o más allá (de un mismo lado de la línea central)
- Secuencia de 15 puntos en C (arriba y debajo de la línea central)
- Secuencia de 8 puntos fuera de las zonas C
Algunos laboratorios deciden aplicar las reglas resaltadas en letra Bold. Otros utilizan las siguientes 3 reglas:
- Un resultado fuera del límite de control superior o inferior (regla 1)
- Dos de cada tres resultados por encima o por debajo del límite de control 2/3 (regla 5)
- Seis resultados en una fila aumentando o disminuyendo (regla 3).
Un conocimiento amplio del comportamiento del proceso podría ser una justificación para el uso de otras reglas o una reducción de las reglas recomendadas.
Construcción de una gráfica o carta de control
Las cartas consisten de un eje X y un eje Y, donde el eje X representa la secuencia del número de lotes en orden adecuado, por ej. Fecha de manufactura o número de análisis.
El eje Y representa los valores de las muestras testeadas, valores promedios, individuales o rangos.
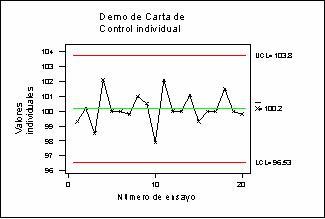
Hay diferentes cartas de control, pero vamos a mencionar la carta de Xmedia / rango muestra el promedio y el rango de un número de valores dentro del lote, por ejemplo para los datos de uniformidad de contenido.
Si hay solo un valor representando el CQA para un lote, la carta preferida es individual / rango móvil, la cual muestra los valores individuales de cada lote y el movimiento del rango para lotes consecutivos. Por ejemplo este tipo de carta puede ser utilizado para valoración de la sustancia activa de lotes de productos.
Establecimiento de los límites de control para tendencia manual
Los datos para ser usados para calcular los límites de control deben ser los datos más recientes que puedan ser considerados bajo control estadístico. Los datos bajo control estadístico no muestran signos de tendencias para arriba o para abajo o cambios en el nivel promedio, ni cualquier signo de valores extremos o diferencias en la variabilidad entre lotes.
Ingresar todos los datos en una carta de comportamiento de proceso y buscar las violaciones a las reglas WEP puede mostrar si los datos están bajo control. Si un solo valor extremo está presente y por otra parte datos en control, este valor puede ser excluido, siempre que tenga una causa asignable y los límites de control ser calculados a partir de los datos remanentes.
Para el uso de la carta individual / rango móvil y el cálculo de los límites de control, se presume que los datos de los Atributos críticos de calidad están normalmente distribuidos. Si los datos no están normalmente distribuidos, estos límites no son apropiados. Ejemplos de CQAs que están normalmente distribuidos son las valoraciones, el promedio de la uniformidad de contenido y el pH, entre otros. Ejemplos de CQAs que no están normalmente distribuidos incluye productos de degradación y valores de aceptación de la uniformidad del contenido.
Test para normalidad pueden ser encontrados en algunos softwares (Bases estadísticas).
Cuando los datos no están normalmente distribuidos, otros enfoques están disponibles para determinar los límites, por ejemplo: límites basados sobre los percentiles de los datos. Estos enfoques típicamente requieren un largo n° de lotes para calcular los límites. (Bases estadísticas).
Espero que les resulte útil, para aquellos que estén interesados en las fórmulas para calcular los límites de las cartas (Xmedia / rango, Individuales /rango móvil), consúltenos en info@cgmpdoc.com.
Para los interesados en profundizar sobre el tema, les ofrecemos un taller sobre Análisis de tendencia de datos modalidad “In Company”, consúltenos en info@cgmpdoc.com.