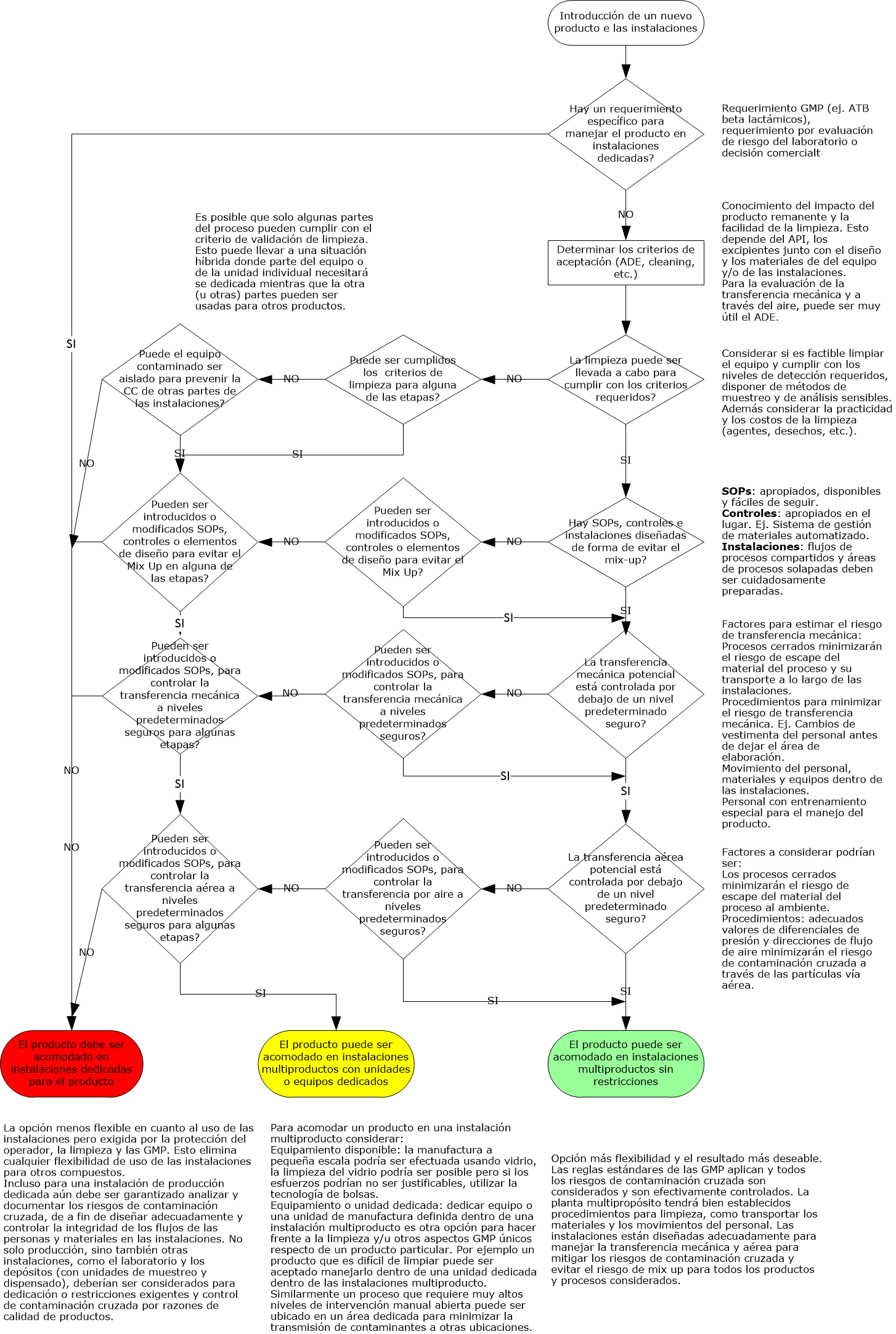
Los requisitos para la validación de la limpieza no están muy claramente definidos en las normas GMP de EE. UU. 21 CFR 210/211. Incluso una Guía de inspección de la década de 1990 sobre este tema no representa la interpretación actual de la FDA. Las cartas de advertencia actuales sobre el tema de la validación de la limpieza pueden ayudar, como la emitida a un fabricante de medicamentos coreano de productos de venta libre.
Requisitos de la FDA en términos de validación de limpieza
La FDA criticó la falta total de una validación de limpieza, a pesar de que se utilizan equipos polivalentes en los que también se fabrican productos no farmacéuticos. La FDA señala explícitamente que los residuos químicos y microbiológicos pueden generar problemas con los medicamentos fabricados en estas instalaciones. Ahora se exige un programa de validación de limpieza correspondiente para todos los productos exportados a los EE. UU., nombrando las condiciones del “peor de los casos”. Los siguientes deben ser considerados como “los peores casos:
* Medicamentos con toxicidades más altas
* Medicamentos con mayor contenido de principio activo
* Medicamentos con baja solubilidad en el reactivo de limpieza
* Medicamentos con propiedades que los hacen difíciles de limpiar
* Limpie los sitios de muestreo en los lugares que son más difíciles de limpiar
* Tiempo de espera antes de limpiar
* Descripción general de los SOP actualizados para garantizar que se implemente un programa adecuado de verificación y validación del proceso de limpieza para productos, procesos y equipos.
Además, la FDA exige que se tomen las medidas necesarias en el sistema de gestión de cambios antes de que se pueda introducir un nuevo equipo o producto de fabricación.
También se puede encontrar más información en la Warning Letter de la FDA a Cosmo Bio Co. Ltd.
Publicado en la News letter GMP de la ECA (Febrero/2022)
 Bueno, pero afortunadamente en la actualidad hay disponibles sistemas de gestión de documentos o DMS (Document Management System).
Bueno, pero afortunadamente en la actualidad hay disponibles sistemas de gestión de documentos o DMS (Document Management System).