La revisión de los BRs de producción junto con la tendencia de los rendimientos y los desvíos son partes claves del proceso de QA para la elaboración de productos.
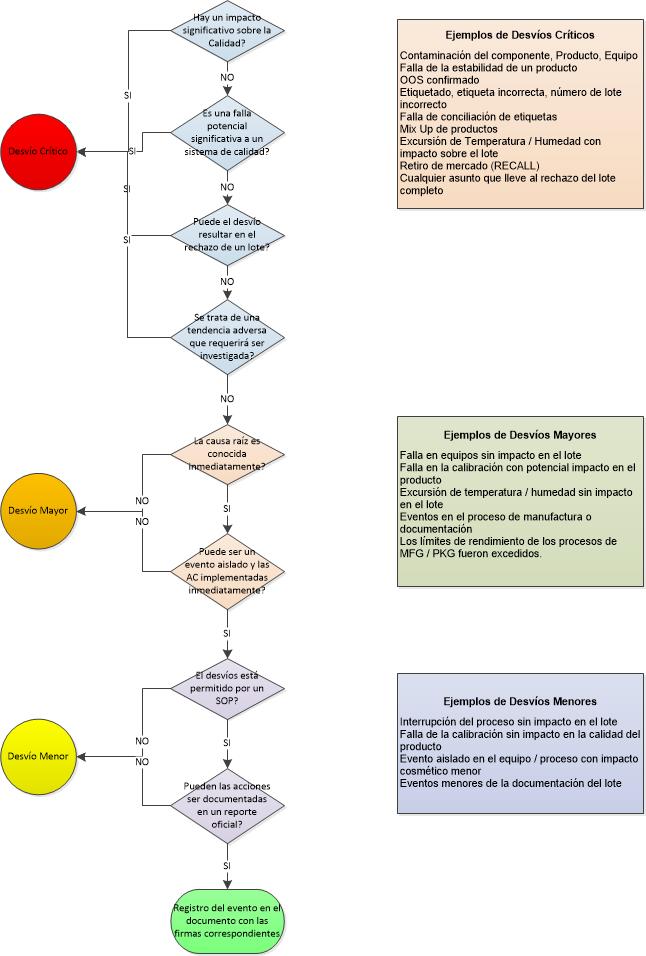
La revisión de BRs es típicamente una etapa de verificación que confirma la aceptabilidad del proceso de elaboración y empaque del producto. Si, de la revisión surge un desvío, el laboratorio gana información de valor desde la investigación que puede llevar a un CAPA y a la mejora del proceso.
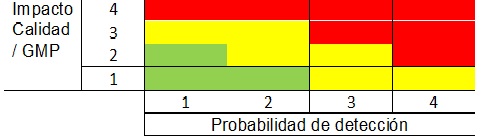
Estas situaciones pueden ser detectadas antes que el rendimiento u otro parámetro exceda los límites de alerta o acción.
Debido a que es un proceso muy común, esta importancia puede ser pasada por alto más allá de los requerimientos regulatorios de las cGMP. Sin embargo la importancia de los registros está relacionada con el respaldo de la elaboración de los productos terminados.
Los requerimientos claves para el registro de la producción y control son encontrados en la subparte J del 21 CFR parte 211, la cual es una de las secciones más largas de la regulación cGMP de la FDA.

Hay un requerimiento cGMP para la confección de Master BR (21 CFR 211.186) identificando las etapas de elaboración / empaque de los productos. El MBR es usualmente la culminación de la transferencia tecnológica, scale up, lotes de validación, y la verificación de las etapas de manufactura alineadas con los requerimientos regulatorios.
El Batch Record, también llamado Batch Production Record o registro de producción del lote, es un documento que confirma el procedimiento paso a paso que los operadores o técnicos deben seguir para elaborar el producto.
Según el 21 CFR 211.188 “Un registro de producción y control de la producción del lote debe ser preparado para cada lote de producto producido y debe incluir información completa relacionada a la producción y control de cada lote”.
Cada BR es un documento controlado desde que es emitido hasta que puede ser eventualmente destruido.
Contiene la misma información que el master BR porque es una reproducción exacta del mismo que ha sido chequeado para su exactitud, fechado y firmado.
El registro del lote será ejecutado por varios operadores para documentar que cada etapa significativa en la manufactura, proceso, empaque o holding del lote fue cumplido.
Esto incluye entre otras cosas, lo siguiente:
- Fechas; cualquier firma además requiere tiempos
- Identidad de equipos individuales mayores y líneas utilizadas
- Identificación específica del lote de componente o material en proceso usado
- Pesos y mediciones de componentes usados durante el proceso
- Resultados de los controles en proceso y del laboratorio
- Inspección del área de elaboración / empaque / rotulado antes de su uso
- Una declaración del rendimiento actual y una declaración del porcentaje de rendimiento teórico para cada fase apropiada del proceso
- Registros de control de rotulados completos, incluyendo muestras o copias de todos los rótulos usados
- Descripción de los contenedores de producto y sus cierres / precintos
- Todo muestreo efectuado
- La identificación de las personas que efectuaron y supervisaron directamente o chequearon cada etapa significativa en la operación
- Cualquier investigación efectuada de acuerdo al 211.192
- Los resultados de las examinaciones hechas de acuerdo con el 211.134
Es destacado enfatizar que el “control” es el corazón de las cGMP y este control extiende a cada etapa crítica en el proceso de manufactura y empaque.
El BR para cada lote es una extensión de este control porque no es suficiente para liberar un producto para la venta hacerlo solo a partir de los resultados analíticos. Los datos de laboratorio deben confirmar los resultados esperados del proceso.
Cada lote ha sido elaborado con controles en proceso. El BR provee una documentación etapa por etapa de esos controles de la manufactura.
El BR completo se convierte en la verificación que todas las etapas críticas fueron efectuadas como estaban descriptas y cuando se combina con los resultados del laboratorio, la firma elaboradora tiene la documentación adecuada para efectuar la disposición del lote.
No nos olvidemos que en paralelo a estos requerimientos de las cGMP tenemos los de las BP de documentación.
Fundamentos del proceso de revisión:
Una vez que el proceso de empaque ha sido completado y que los registros han sido devueltos a la unidad de calidad quien registra tales documentos, el paso siguiente es la revisión de los BR para exactitud y conformidad de los estándares establecidos en los documentos.
Es recomendable, es más diría que hoy es un requerimiento, que sea efectuada una revisión del BR de manufactura o empaque por parte del equipo de manufactura o personal de empaque antes de regresar el registro del lote al departamento de QA.
El requerimiento regulatorio para BRR es hallado en el 21 CFR 211.192 y declara: “todo registro de producción y control de producto, incluyendo aquellos de empaque y rotulado, deben ser revisados y aprobados por la unidad de control de calidad para determinar su cumplimiento con todo lo establecido, aprobado en procedimientos escritos antes que el lote sea liberado o distribuido. Cualquier discrepancia no explicada (incluyendo un porcentaje de rendimiento teórico que excede el máximo o mínimo establecido en el Maestro de producción y los registros de control) o la falla de un lote o cualquiera de sus componentes para cumplir con cualquiera de sus especificaciones debería ser exhaustivamente investigada, haya sido o no distribuido el lote. La investigación debe ser extendida a otros lotes del mismo producto e incluso a otros productos que hayan podido ser afectados por la falla o discrepancia. Un registro escrito de la investigación debe ser efectuado y debe incluir conclusiones y seguimiento”.
Cada BR es típicamente específico para el producto y potencia del producto porque contiene las cantidades de API y excipientes requeridos. En el caso de comprimidos el Br puede indicar por medio de imagen o foto, o un dibujo, la descripción del tamaño, forma, color y cualquier estampado en relieve sobre el comprimido. Si el comprimido es recubierto, las letras sobre el comprimido y el color puede ser registrado.
En el caso de líquidos orales, pueden ser descriptos su color y aroma. Estos detalles pueden ser parte del MBR.
Así como la elaboración de productos es guiada por procedimientos y por los BR, la revisión del BR debe ser manejada a través de un SOP. A pesar que la revisión del BR puede ser menos complicada que la elaboración y empaque de productos, un SOP debería hacer que el proceso sea más consistente y disponer además de un check list para la revisión asegurará que las etapas y parámetros críticos sean revisados.
En un segundo artículo hablaremos sobre el SOP de BRR (revisión de BR) y su check list, correcciones de BR y algunas inconsistencias en la revisión de BRs.