Si miramos las observaciones de las agencias como la FDA
y la EMA, la validación de procesos y la calificación de equipos están dentro
del top ten de deficiencias encontradas. Además de deficiencias relacionadas
con el diseño de locales y equipos, y pobres instrucciones sobre calibración, limpieza
y mantenimiento de los equipos.
¿Cómo podemos llevar a cabo
una calificación que cumpla con las cGMP y, además sea eficiente?
Matthias Klein de CSL-Behring, reconoce que la clave del
éxito radica en el uso eficiente de los documentos y procesos existentes
(incluidos los sistemas de control de cambio y calibración) en las compañías
farmacéuticas. La minimización del gasto también es posible aplicando de manera
consistente las GPE de acuerdo con la Guía de referencia del ISPE sobre commissioning
y calificación como un concepto de calificación integrado. Podemos mencionar los
documentos de pruebas de aceptación de fábrica (FAT) o las pruebas de
aceptación del sitio (SAT). Estas pruebas efectuadas por personal capacitado, y
documentadas adecuadamente, nos permitirá evitar la duplicación de trabajo.
Para una efectuar una calificación eficiente y pragmática
debemos usar el análisis de riesgos del proceso de manera apropiada (la ICH Q9
da los lineamientos y una serie de herramientas, entre las que mencionamos el FMEA).
El análisis de riesgos apropiado reduce aún más el
esfuerzo de calificación.
Un análisis de riesgo general es la base para la
calificación de diseño (DQ). Los componentes relevantes para la calificación son
identificados y el grado de la calificación se determina mediante un análisis
detallado del riesgo. El análisis de riesgo también debe considerarse durante
el curso de la calificación y en la operación de rutina.
Durante el proyecto de calificación, el control de
cambios se puede manejar de manera casi pragmática.
Las razones para las pruebas de calificación se
cuestionan cada vez más en el contexto de las inspecciones y auditorías en las
que el concepto integrador y una matriz de trazabilidad adecuada pueden
garantizar que solo se realicen las pruebas que provienen del análisis de
riesgos.
En cuanto a la recalificación ahora se requiere una
evaluación periódica de los locales, sistemas, equipos y servicios en relación
con el estado de calificación. También se deben evaluar pequeños cambios. La
frecuencia de esta evaluación debe ser justificada.
La Guía de la FDA ya no contiene el término revalidación.
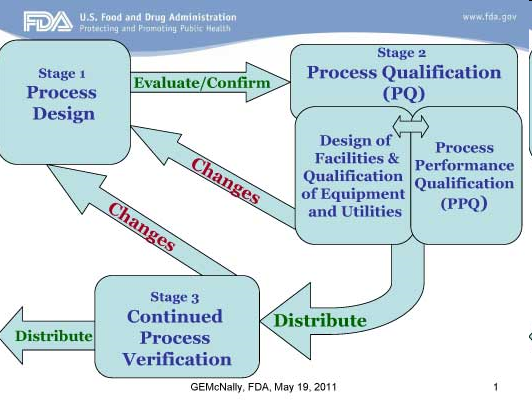
Con el enfoque del ciclo de vida, el tema continúa en la etapa 3 Verificación
continua del proceso, cuyo objetivo es mostrar que el proceso permanece en un
estado validado durante la producción de rutina. Para este propósito, se solicita
un sistema que detecte desviaciones de proceso no planificadas. El proceso no
debe salirse de control. Aquí se hace una referencia directa a la Revisión
Anual del Producto (21 CFR 211.180) para apoyar este programa en curso. Y se
atribuye gran importancia a las estadísticas.
¿Y el futuro?
El ISPE y la ASTM (Sociedad Americana de Pruebas y
Materiales) apuntan a una “calificación de valor agregado”. Debemos
enfocarnos en las especificaciones de requisitos del usuario (URS) y la
Calificación de proceso (PQ).
El ISPE resume los “Principios para la Calificación
del Siglo 21” en diez puntos, que es un excelente resumen de los
principios descritos en la norma ASTM E2500:
- Enfoque
en la calidad del producto.
- “Requisitos
del usuario” basados en el proceso. Se confirman como satisfechos en el PQ
(IQ y OQ están subordinados en importancia).
- “Evaluaciones
de riesgo”: el desarrollo del proceso y el diseño experimental son los
elementos clave para identificar funciones y parámetros críticos.
- Solo
se utilizarán los parámetros críticos del proceso como base para la
calificación.
- Todas
las actividades deben aportar valor al proceso (“No haremos nada solo por
el cumplimiento de la normativa”).
- Actividades
de calificación basadas en el riesgo. Ej. clasificación GAMP.
- “Documentos
de valor agregado”.
- Uso
de la documentación del proveedor, si es posible.
- Pruebas:
por regla general, deben realizarse una sola vez. Pero puede ser necesario
repetir algunas pruebas que se han llevado a cabo en una etapa anterior del
desarrollo.
- Fomento de la innovación: se requiere
la flexibilidad de los programas de calificación para poder implementar nuevas
tendencias.
Todo esto requeriría grandes cambios organizativos en las
empresas (probablemente uno de los mayores obstáculos) que a menudo tienen
conceptos relativamente rígidos de calificación y validación.
El riesgo para la calidad del producto se deberá basar en
el conocimiento del proceso.
Este proceso tiene lugar bajo el paraguas de las Buenas
Prácticas de Ingeniería y sobre la base de la gestión de riesgos, la gestión de
cambios y la revisión del diseño. El input al proceso es el conocimiento del
producto, el conocimiento del proceso, los requisitos reglamentarios y los
requisitos de calidad de la empresa.
El output esperado, es la operación y la mejora continua.
En la Guía de Validación de Procesos de la FDA, las
actividades de calificación son parte de la Etapa 2 del ciclo de vida de
validación del proceso de calificación del proceso. Los términos DQ, IQ y OQ ya
no se usan en el documento. Esto es consistente con la Guía Estándar ASTM E2500
y GAMP 5. Ambos documentos se abstienen de usar estos términos y los sustituyen
por “verificación”. La intención es dejar claro que las prácticas de
la industria de DQ, IQ y OQ, etc. no son un requisito reglamentario y fomentar
una aplicación más sólida de las Buenas Prácticas de Ingeniería (GEP).
La revisión del Anexo 15 va en la dirección de las GEP y el
FAT y SAT, y si bien menciona etapas de calificación como DQ, IQ, OQ, PQ las
mismas no son obligatorias.
De acuerdo a la Guía de Validación de Procesos de la FDA
los lotes de la Calificación de Rendimiento del Proceso (PPQ), deben ser
llevados a cabo por empleados de producción calificados utilizando
instalaciones y equipos calificados; bajo las condiciones de fabricación
comerciales y, en última instancia, pueden ser lanzados como lotes comerciales
normales, dependiendo de un resultado de Calificación de Proceso global
exitoso.
De acuerdo a la FDA “Una PPQ exitosa confirmará el
diseño del proceso y demostrará que el proceso de fabricación comercial
funciona como se esperaba”. Por lo tanto, la terminación de PQ antes de la
comercialización es obligatoria.
La FDA considera PPQ en el sentido del documento PIC / S
PI 006. Ha igualado PQ y la validación de procesos desde 1996. En el Anexo 15
revisado, PQ y Process Validation se pueden combinar.
Conclusión
El futuro de la calificación parece residir en una
calificación aún más integrada que involucra a ingenieros y calificadores, en
un proyecto estructurado basado en el análisis de riesgos e involucrando la
mayor cantidad posible de documentos GEP. Las pruebas de los preceptos URS en
el PQ serán la base para las principales pruebas de calificación. Seguramente
el nuevo modelo de “verificación” de la norma ASTM E2500 reemplazará las
etapas de calificación clásicas.
El futuro de la validación estará enfocado en la comprensión
del proceso. Las estadísticas serán útiles para apoyar estos aspectos.
En Europa, el enfoque “tradicional” seguirá
estando disponible como se indica en la Guía de la Validación de Procesos de la
EMA y en el Anexo 15 revisado. Este enfoque tradicional ya no se menciona en la
Guía de la FDA.
¿Qué hay de los productos heredados (productos antiguos)?
La FDA dice que comienza con la etapa 3 “Verificación continua del
proceso”. En el Anexo 15 revisado no hay pistas sobre productos heredados,
aunque el enlace a PQR indica que los productos heredados también están en el
foco de la verificación del proceso en curso. El enfoque de EMA es muy intensivo en la
“verificación continua del proceso” ahora, más que en la Guía de la
FDA.
Hasta ahora, a menudo se consideraba que la PQ estaba
relacionada principalmente con el equipo. La perspectiva cambió en la guía de
la FDA, con la nueva expresión PPQ como parte de PQ (ahora conocida como Calificación
de rendimiento del proceso).
Tomado de: ECA
Validation Good Practice Guide