Hay con frecuencia muchas discusiones en la industria farmacéutica respecto de la naturaleza exacta del proceso de validación. La validación de proceso es un requerimiento de las cGMP y por lo tanto aplica a la elaboración de productos medicinales y dispositivos médicos.
La FDA define validación de procesos como:“establecer evidencia documentada, la cual provee un alto grado de seguridad que un proceso específico producirá consistentemente un producto que cumpla con especificaciones predeterminadas y características de calidad”.
La intención original de la validación fue clara y simple, evidencia documentada que un proceso hace lo que pretende hacer y es exitoso como proceso. Actualmente Plan Maestro de Validación formal es requerido por la mayoría de empresas y este documento contiene enormes detalles, muchos de los cuales son duplicados en los documentos de IQ, OQ y PQ, y los costos de validación en la mayoría de los proyectos superan el 10 % del costo total del proyecto.
La verdadera intención de la validación, tal como está definido en la guía de la FDA, puede haber sido perdida en la interpretación que la industria que tiene que testear 3 lotes con resultados aceptables. Estos tres lotes son probablemente los que tienen menos variación respecto de cualquier de los lotes producidos en la planta, debido a la necesidad crítica de completar la validación de manera exitosa, y los cuidados de la manufactura en esos tres lotes de validación.
Sin embargo, para obtener el conocimiento necesario de los productos y procesos, y al mismo tiempo producir un retorno de la inversión más atractiva, la validación debe basarse en la ciencia.
Si un modelo DOE se utiliza tempranamente en el proceso para caracterizar el producto y el proceso, el modelo confirmado puede luego ser usado para establecer límites óptimos para maximizar el resultado y minimizar la variación y predecir que podría suceder bajo las condiciones alternativas.
Posts tagged ‘Plan Maestro de Validación’
La nueva disposición de la ANMAT tiene establecidos requerimientos para los sistemas computarizados o informatizados en el Anexo 6 Sistemas Informatizados de la nueva Disposición.
Este Anexo aplica a todas las formas de sistemas informatizados usados como parte de las actividades reguladas por las BPF y se utilicen para crear, modificar, mantener, archivar, obtener o distribuir registros electrónicos.
Además de Validar las aplicaciones, debemos calificar la Infraestructura informatizada (IT).
La inclusión de un sistema informatizado en la manufactura no debe aumentar el riesgo total del proceso.
Estos lineamientos de la nueva GMP están basados en la GAMP5 (ISPE) A Risk-based Approach to Compliant GxP Computarized Systems.
Como parte del sistema de gestión de riesgos, las decisiones sobre la extensión de la validación y de los controles de la integridad de datos deben basarse en una evaluación de riesgos del sistema informatizado justificada y documentada.
La norma menciona diferentes figuras que deben existir, como: el propietario del proceso (process owner), el propietario del sistema (system owner), las Personas Calificadas e IT. Obviamente todo el personal debe estar calificado, con su nivel de acceso y definidas sus responsabilidades.
Los proveedores de sistemas informatizados deben estar evaluados y debe disponerse de acuerdos técnicos y acuerdos de confidencialidad (ppalmente. para acceso remoto). La necesidad de auditarlos debe estar basada en un análisis de riesgo del mismo.
Debe disponerse de un inventario de los sistemas computarizados del laboratorio.
La Validación debe ser proporcional al riesgo del sistema (clasificación del riesgo, por ejemplo Categoría GAMP5 y calificación del proveedor), a más riesgo más esfuerzos de validación.
La transferencia de datos debe ser efectuada de forma segura.
En cuanto a la Operación de los SC, debemos asegurar que los datos son guardados regularmente de forma integra durante el período de conservación de los mismos. Debe haber un Audit Trail y debe ser considerada la Gestión de cambios y configuración.
Los sistemas informatizados deben ser revisados periódicamente para verificar la validez de la validación. Esta revisión incluye por ejemplo documentos de operación, validación, cambios, eventos, accesos y manejo de back up, por ej.
También hace referencia a otros temas de importancia como el manejo de firmas electrónicas, plan de Disaster Recovery, etc.
Desde cGMPdoc les ofrecemos distintas alternativas como:
- Entrenamientos “In Company” para capacitar a su personal
- La implementación del proceso de CSV (Validación de Sistemas Computarizados), lo cual incluye la discusión del procesos internamente con el laboratorio, la confección de los SOPs necesarios, el entrenamiento del personal, la confección del PMV de sistemas computarizados y la ejecución de una validación a modo de ejemplo, de manera que Uds. puedan luego continuar con las actividades del Plan.
- La validación de un sistema computarizado
- Asesoramiento en la revisión de su proceso de CSV si lo necesita
- Pack de SOPs y documentos modelos sobre CSV
Espero que les resulte de utilidad.
Si bien algunas regulaciones no exigen un Plan Maestro de Validación (PMV) formal, como por ej. el Código Federal de Regulaciones (CFR) en USA, en nuestro País la Disposición 3827/2018 sí lo exige, y suele ser uno de los primeros documentos que los inspectores solicitan al auditar el laboratorio.
El PMV es una herramienta muy útil para documentar la forma en que las actividades de Validación son manejadas en el laboratorio.
Inicialmente el PMV debería definir la Política de Validación, la organización y el alcance de las actividades.
Podemos dividir al PMV en 3 principales secciones:
- Inventario de los sistemas, considerando equipos, procesos, áreas, métodos, etc. Indicando cuáles de ellos deben ser validados y el nivel de validación de cada uno.
- Agenda de las actividades de validación, la cual nos da el orden de acuerdo a prioridades, obviamente no podemos validar un proceso sino tenemos calificados los equipos que son utilizados en dicho proceso.
- Actividades de mantenimiento del estado validado, se trata de las actividades de monitoreo de la performance de los sistemas. Un ejemplo de esto es confeccionar un reporte anual de monitoreo ambiental para zonas limpias validadas.
Además es importante mencionar en el PMV las referencias a las regulaciones que lo rigen y los procedimientos y documentos (protocolos y reportes) que serán escritos para cumplir con los requerimientos del PMV.
En el caso que un laboratorio que ya ha iniciado las actividades de validación y NO tiene un PMV, es recomendable escribir uno. Este PMV retrospectivo sería esencialmente un resumen formal de las actividades que están ejecutadas , como fueron ejecutadas y además listar las actividades pendientes.
La estrategia y el contenido del PMV refleja la aptitud de la organización para la Validación. El tiempo invertido en la confección de un PMV reporta en beneficios cuando el mismo es solicitado por los inspectores de las agencias regulatorias.
Estas líneas son una guía para efectuar la calificación de las instalaciones, equipos y servicios existentes.
Existen guías que aportan mucho detalle para la calificación de los equipos nuevos, sin embargo debemos reconocer que cuando nos referimos a la calificación del equipamiento existente, la información es limitada.
En este artículo queremos dejarles reflejado nuestro pensamiento sobre este tema.
No es inusual encontrar que una planta tiene una mezcla de equipos nuevos calificados completamente (DQ, IQ, OQ, PQ) y equipamiento parcialmente o NO calificado.
A través de muchos años de uso, las instalaciones, servicios, sistemas y equipos han mostrado su aptitud para funcionar de acuerdo a distintos requerimientos.
Sin embargo, las cGMPs necesitan que los ítems que tienen impacto sobre la calidad del producto, proceso deben estar calificados. La calificación formal es la base de las actividades relacionadas tales como la validación asociada con la introducción de nuevos productos o la validación de los productos existentes que forma el punto de partida para futuros controles de cambio.
La calificación debería proveer verificación documentada que los parámetros definidos como críticos para la operación y mantenimiento son adecuadamente controlados.
Es esencial que la calificación sea práctica y alcanzable, agregue valor al proyecto y esté concentrada en los elementos críticos del equipo. Es recomendable que un enfoque de Análisis de Riesgos sea usado para definir la profundidad de la calificación.
Siempre debería haber una proporción razonable entre el riesgo para la calidad del producto, la cantidad de mediciones a ser efectuadas y la documentación a ser preparada. Por ejemplo la etapa de manufactura, estado regulatorio y el propósito de uso.
Si miramos los requerimientos regulatorios, nos encontramos que la ICH Q7a indica que las Instalaciones, servicios, equipos y sistemas deben ser adecuadamente calificados para asegurar la integridad de los datos y del producto.
Un lineamiento adicional esta dado por las PIC/S (Pharmaceutical Inspection Co-operation Scheme), donde las mismas indican que aunque no sea posible realizar los detalles de una calificación de la instalación para un equipo establecido ni el enfoque detallado para una calificación de la operación, debería haber datos disponibles que verifique y soporten los parámetros operativos y los límites para las variables críticas de la operación del equipamiento.
Adicionalmente, la calibración, limpieza y mantenimiento preventivo, deberían estar documentados y por supuesto deben existir SOPs de operación y procedimientos de entrenamiento de los operadores para el uso del equipamiento.
Ahora vamos a ver el análisis de riesgo (AR) aplicado a las actividades de calificación.
El AR es un enfoque formal y sistemático para identificar riesgos GMP relacionados al equipamiento y sistemas de soporte. Es una herramienta muy útil que puede ser aplicada a la planta, equipamiento y sistemas los cuales han estado en uso por muchos años.
Es recomendado que las instrucciones de manufactura sean usadas en combinación con los requerimientos generales GMP (ej. Diseño, documentación de riesgos de contaminación, utilización en el producto final) como base para el AR.
Cada etapa de la manufactura puede ser evaluada individualmente con respecto a operaciones / actividades críticas (por ej. Velocidad de agitación, rangos de temperatura, presión, humedad, manejo de producto expuesto, etc.).
Descripciones de procesos, reportes de desarrollo y revisiones de calidad del producto pueden asistir en la identificación de las operaciones parámetros críticos de calidad.
El AR puede ser llevado a cabo usando distintos enfoques, ej. HACCP, FMEA, árbol de decisiones, etc. el AR del equipamiento puede ser efectuado para el tren de equipos o por unidad de proceso individual (ej. Reactor, centrífuga, etc.) y sus sistemas de soporte.
Las unidades de operación podrían ser evaluadas de acuerdo al siguiente esquema:
- La operación impacta directamente sobre la calidad del producto?
- La operación crea datos electrónicos los cuales son la base para actividad GMP relacionada?
- Un malfuncionamiento podría impactar directamente sobre la calidad del producto?
- Tiene instrumentos que miden o controlan etapas críticas del proceso?
- La operación / instalación causa riesgo de contaminación al producto o al ambiente de la planta?
- Los materiales de construcción están en contacto directo con el producto?
Si al final de las preguntas anteriores nos encontramos con algún “Si” entonces la operación es considerada como GMP relevante.
Durante el AR, debemos considerar la probabilidad de ocurrencia y la detectabilidad y las medidas para reducir el riesgo deben ser identificadas.
El AR puede ser efectuado usando un template, donde las etapas de trabajo típicas o actividades para la unidad del proceso son listadas (incluyendo instrumentos y sistemas de control).
Adicionalmente los materiales de construcción en contacto directo con el producto son evaluados y los dispositivos de medición y control críticos son identificados para su calibración.
Ahora veamos las actividades de calificación, en el siguiente flujograma:
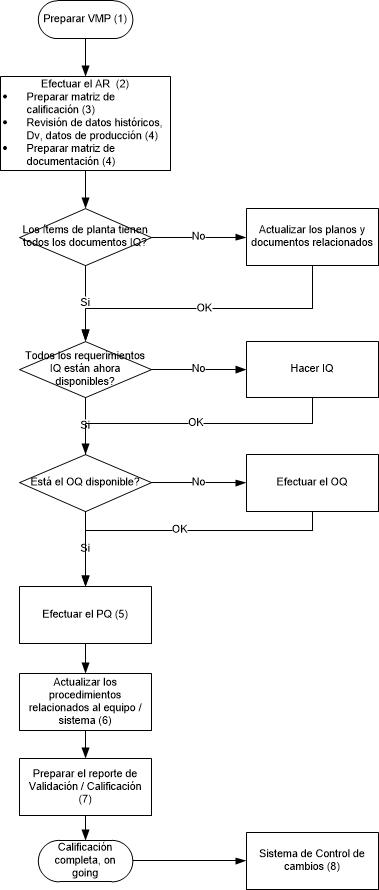
Adicionalmente, los equipos que son calificados, dependiendo de la criticidad de los mismos pueden ser ingresados en un plan de revisión periódica.
El objetivo de este documento es brindar una idea de los elementos que son requeridos para la confección de un Plan Maestro de Validación.
Esta guía intenta mostrar un modelo de listado de items, pudiendo ser utilizado otro orden e incluso algunos elementos más u otros menos.
1. Logo del Laboratorio
2. Breve resumen de la empresa
3. Organigrama de la empresa
4. Responsabilidades (distribución de las mismas)
5. Planos del laboratorio (Lay out, flujo de personal, flujo de materiales, etc.)
6. Servicios utilizados (agua purificada, aire comprimido, nitrógeno, vacío, sistemas de extracción de polvos, HVAC, etc.)
7. Listado de productos (código, nombre de los productos, forma farmacéutica, principio activo, concentración, tamaño del lote, cantidad de lotes anuales), de su análisis surge la elección de los productos a validar (*)
(*) Nota importante: los productos que presentan problemas de elaboración no deben ser incluidos dentro de las actividades de validación, hasta tanto los problemas de dichos productos no sean solucionados
8. Hoja de seguridad de los principios activos o sus DL50, solubilidad y mínima dosis diaria, en el caso de disponer de dicha información sin la correspondiente hoja de seguridad, es importante hacer referencia a la fuente.
Con la información de la tabla podrá ser calcula un Factor que permitirá ordenar a los activos por su criticidad y determinar el peor caso (worst case) para las actividades de Validación de limpieza.
9. Listado de áreas (productivas y de control).
10. Listado de equipos (productivos y de control) con su codificación y sector.
11. Listado de Instrumentos, Plan de calibración, frecuencia de calibración de los mismos, indicando si es externa o interna.
12. Diagrama de flujo de elaboraciones por los trenes de elaboración, indicar para cada flujo de proceso aquellos productos que pasan por dicho tren. Por ej.: Molino / Tamiz /Mezclador doble cono/ Comprimidora / Paila de recubrimiento.
Para productos que tienen 1 o más etapas de proceso en terceros indicar: producto, etapa tercero
13. Listado de métodos analíticos (indicar el estatus de validación de los mismos)
14. Listado de métodos analíticos de trazas (si disponen de los mismos)
15. Listado de SOPs (indicando código, versión, título del documento, fecha de vencimiento)
16. Plan de mantenimiento preventivo
17. SOP de Control de cambios
18. SOP de Revisión Anual de Productos (APR)
19. Plan capacitación
20. SOP de Revalidación
Una vez disponible la información anterior y habiéndose efectuado los análisis de riesgos correspondientes se procede a:
- Elaborar el documento de Plan Maestro de Validación propiamente dicho, donde además de todos los inventarios e información solicitada anteriormente, se documentan los criterios de aceptación para las distintas actividades, como por ejemplo cantidad de lotes consecutivos exitosos para la validación de procesos, o cleaning, etc.
- Se elabora el gantt de actividades, se trata de la agenda donde se programan las tareas, con fechas y responsables de efectuarlas. Las actividades deben estar coordinadas de tal manera que para la validación de un proceso, previamente se hayan efectuados las calificaciones correspondientes de servicios, áreas, equipos a utilizar en la elaboración del mismo, así como también la metodología de análisis y control debe estar validada. Generalmente los gantts de trabajos son elaborados con12 a24 meses de horizonte y se van actualizando.

Suelo caminar por los pasillos de algunos laboratorios y en muchos casos escucho a la gente desconforme con las actividades de validación.

Algunos de los comentarios más oídos son sobre:
- La duración de los proyectos y lo difícil que les resulta cumplir con la agenda de los mismos
- La falta de personal calificado o especializado en Validación
- Los circuitos de revisión / aprobación de los documentos confeccionados, la mayoría piensan que son demasiados los que participan de los circuitos, lo cual los hacen lentos y burocráticos
- La falta de disponibilidad de los equipos, servicios o áreas tanto de producción como del laboratorio, los planes de validación son relegados por un tema de prioridades. Recuerdo que a veces nos pasaba que teníamos las muestras para efectuar determinaciones de una validación de un proceso, sin embargo la carga del laboratorio era tal que no nos daba lugar a analizarlas, ergo: las muestras envejecían y el plan se demoraba por un tema claramente entendible de prioridades.
- Por último y para no cansarlos, análisis de riesgo, pero, que es esto? Muchos no comprenden claramente que es lo que deben validar y finalmente validan todo.
La idea por la cual comenzamos a pensar en este artículo, es para que sirva de inicio de una discusión sobre este tema y como Uds. son los expertos en el tema, queremos aprovechar la oportunidad de aprender de todos.
Todas sus opiniones y experiencias son bienvenidas!!!