Las Buenas Prácticas de Manufactura (GMP) en la industria farmacéutica están diseñadas para garantizar que los productos se produzcan y controlen consistentemente de acuerdo con estándares de calidad definidos. Sin embargo, es común que las empresas sobre-interpreten las regulaciones, lo que lleva a procesos innecesarios que pueden inflar los costos y reducir la eficiencia. Para ajustar las GMP y el cumplimiento, y avanzar hacia sistemas GMP eficientes sin dejar de cumplir con las regulaciones, las empresas pueden considerar varias estrategias:
Enfoque basado en riesgos: implementar un enfoque basado en riesgos para la gestión de calidad, donde el enfoque está en los procesos y áreas que tienen el impacto más significativo en la calidad del producto y la seguridad del paciente. Esto permite a las empresas asignar recursos de manera más efectiva y priorizar áreas que requieren un cumplimiento estricto, reduciendo controles innecesarios en áreas de menor riesgo.
Mejora continua: adopte metodologías de mejora continua como Six Sigma o Lean Manufacturing para agilizar los procesos, eliminar desperdicios y mejorar la eficiencia. Estas metodologías pueden ayudar a identificar actividades que no agregan valor y reducir la complejidad en los sistemas GMP.
Capacitación y compromiso de los empleados: asegúrese de que los empleados estén bien capacitados y comprendan la intención detrás de las regulaciones GMP. Una fuerza laboral informada puede tomar mejores decisiones sobre lo que es necesario para el cumplimiento y lo que podría ser una sobreinterpretación de las reglas.
Simplifique la documentación: revise y simplifique los procesos de documentación. Si bien la documentación es un aspecto crítico de las GMP, una documentación demasiado complicada o excesiva puede resultar contraproducente. Asegúrese de que los documentos sean claros, concisos y solo lo detallados que sean necesarios para cumplir con los requisitos de cumplimiento.
Utilice tecnología: aproveche la tecnología y la automatización para reducir los errores manuales y mejorar la eficiencia. Las soluciones digitales pueden ayudar a gestionar la documentación, realizar un seguimiento del cumplimiento y optimizar los procesos de gestión de calidad.
Evaluación comparativa y mejores prácticas: consulte las evaluaciones comparativas y las mejores prácticas de la industria para comprender cómo otras empresas cumplen de manera eficiente los requisitos de GMP. Aprender de otros puede proporcionarle información sobre cómo optimizar sus propios procesos.
Diálogo regulatorio: participar en un diálogo abierto con las autoridades regulatorias para obtener una comprensión más clara de las expectativas de cumplimiento.
Subcontratación de actividades complementarias: Considere la posibilidad de subcontratar actividades complementarias a socios especializados que puedan realizarlas de manera más eficiente y de conformidad con las GMP. Esto permite a la empresa centrarse en sus competencias principales y al mismo tiempo garantizar el cumplimiento en todas las áreas de operación.
Al centrarse en estas estrategias, las empresas pueden desarrollar sistemas GMP eficientes que no sólo cumplan con las normas sino que también estén optimizados para lograr eficiencia y eficacia. Se trata de encontrar el equilibrio adecuado entre garantizar la calidad y la seguridad del producto y al mismo tiempo eliminar costos y procesos innecesarios.
El día 16/6/2023 fue publicada en el boletín oficial esta nueva disposición de la ANMAT: Guía de Buenas Prácticas de Fabricación para elaboradores, importadores / exportadores de medicamentos de uso humano, la cual, de acuerdo a lo indicado en la disposición, está en vigencia a partir del 16/7/2023, de acuerdo a lo establecido en el artículo 3.
ARTÍCULO 1°.- Apruébanse los requerimientos denominados “Guía de Buenas Prácticas de Fabricación para Elaboradores, Importadores/Exportadores de Medicamentos de Uso Humano” que como ANEXO DI-2023-64216567-APN-INAME#ANMAT, forman parte integrante de la presente disposición.
ARTÍCULO 2°.- Deróganse las Disposiciones ANMAT N° 2819/04, 3602/18, 3827/18 y 1281/19.
ARTÍCULO 3°.- Establécese que la presente disposición entrará en vigencia a los 30 (treinta) días hábiles de su publicación en el Boletín Oficial.
En otra parte del texto, la disposición indica:
Que las Buenas Prácticas de Fabricación para Elaboradores, Importadores/Exportadores de medicamentos contemplan los lineamientos internacionales aprobados por la Organización Mundial de la Salud, informes de la PIC’S – Pharmaceutical Inspection Cooperation Scheme, como así también por normas de ICH – International Council for Harmonisation – e ISO -International Organization Standarization.
Que como consecuencia de los avances científicos y tecnológicos resulta necesario adoptar nuevos requerimientos internacionales sobre Buenas Prácticas de Fabricación de Especialidades Medicinales, tales como los aprobadas por la Pharmaceutical Inspection Cooperation Scheme (PIC’S) PE 009-16 Parte I y II del 2022, como así también la norma de International Council for Harmonisation ICH Q7A.
Les dejo el link a la Disposición con su anexo correspondiente.
Hoy quiero compartir con Uds, este artículo escrito por Thomas Peither, GMP-Verlag Peither AG
Un sistema CAPA debe entenderse como un elemento importante del sistema de calidad farmacéutica e implementarse de manera uniforme en toda la empresa o grupo. En principio, se puede implementar de dos formas diferentes: como un sistema autónomo o como un sistema integrado. En este artículo, profundizaré en los pros y los contras de cada uno, y también compartiré algunas de las mejores prácticas generales de CAPA.
Sistemas CAPA Autónomos
Un sistema CAPA autónomo se caracteriza por su propia documentación y seguimiento de horarios. Un SOP autónomo describe el proceso para la definición, responsabilidad, seguimiento del cronograma y cierre de las actividades CAPA. Estas se derivan como posibles actividades de seguimiento a incidencias que están reguladas en otros sistemas, como por ejemplo para la atención de desviaciones, queja / reclamos o autoinspecciones.
En base a una queja, por ejemplo, se inicia un proceso CAPA en el sistema CAPA independiente después de su evaluación de riesgos, si es necesario, y se realiza un seguimiento mediante un sistema de documentación separado.
Los sistemas autónomos tienen una ventaja decisiva: se pueden tomar medidas inmediatas para eliminar la desviación y aún se pueden documentar y procesar correcciones rápidas dentro del sistema de activación, evitando así una generación inflacionaria de procesos CAPA.
Otra ventaja de un sistema autónomo es una clara separación entre el sistema CAPA y otros procesos. Por lo tanto, cada uno de los sistemas puede ser monitoreado, documentado y discutido por sí solo.
Sin embargo, esto también conlleva una desventaja: los dos procesos separados deben referenciarse entre sí para vincularlos de manera clara y comprensible. Esto siempre conlleva el riesgo de que la información se pierda o se transfiera incorrectamente.
Además, eventos similares que pueden no ser espectaculares cuando se ven individualmente pueden convertirse en debilidades del sistema cuando se ven como un todo, lo que es más difícil de reconocer cuando se ven por separado.
Los sistemas de IT son, por supuesto, útiles aquí. Cada uno genera procesos específicos para las diferentes actividades, pero están interrelacionados y hacen accesible toda la información del otro proceso.
Sistemas CAPA Integrados
Un sistema CAPA integrado suele ser algo más simple. Esa es una clara ventaja. Aquí, al final del procesamiento del incidente inicial, se definen las medidas CAPA correspondientes en la misma hoja de documentación y posteriormente también se documenta su implementación. Después de manejar tanto el problema como las medidas CAPA, la unidad de calidad responsable puede evaluar y cerrar ambos procesos juntos.
Esta ventaja, por otro lado, hace que el proceso sea más complejo: una vez finalizado todo el proceso, se debe transferir de nuevo a la unidad de calidad para su evaluación estadística (análisis de tendencias, revisión por la dirección, etc.) y cierre en el sistema de gestión de calidad. Si hubiera inquietudes por parte de la unidad de calidad, el sistema debe otorgar a la unidad de calidad un derecho de rechazo. En este caso, el procedimiento será transferido nuevamente a la unidad responsable para lograr una conclusión aceptable.
Aspectos importantes para ambos sistemas CAPA
Autónomo o integrado, para ambas variantes, la descripción del sistema CAPA debe hacerse en el manual de gestión de la calidad junto con la descripción de los sistemas para desviaciones, quejas, retiros, etc.
Además, un sistema CAPA requiere su propio SOP en el que se definen los procedimientos junto con las responsabilidades y la documentación necesaria.
Tiene sentido prescribir un análisis de riesgo posterior a la investigación y procesamiento de las desviaciones y, en consecuencia, la decisión sobre el posible inicio de una actividad CAPA.
No importa a qué resultado se llegue, en todo caso se documenta que se ha pensado en una medida más en el sentido de CAPA.
Tres aspectos son importantes aquí:
Seguimiento rápido de la implementación,
Control de eficacia y
Informar las actividades de CAPA en la revisión por la dirección.
¿Cuáles son las características de un sistema CAPA eficaz?
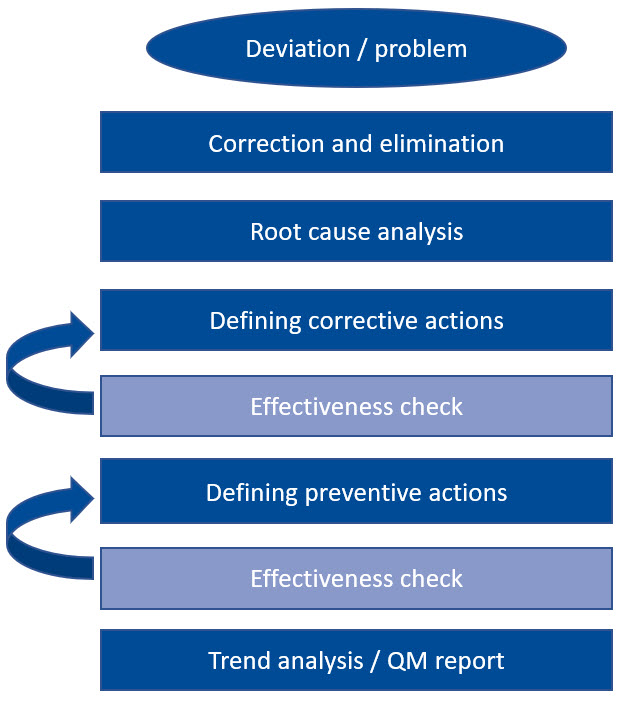
En última instancia, todos los sistemas CAPA efectivos se basan en los mismos procesos. Al definir las medidas CAPA, es importante determinar cómo se puede comprobar su eficacia. Para ello, se necesitan criterios objetivos que puedan utilizarse para determinar el resultado. Si la medida tomada no muestra el éxito esperado, se debe considerar una nueva acción correctiva o preventiva. Por lo tanto, un sistema CAPA es un proceso iterativo continuo que sigue hasta que conduce a un resultado satisfactorio.
Veamos un ejemplo: la primera acción es siempre la corrección y eliminación inmediatas de la desviación, seguida inmediatamente por una investigación de la causa del error. Una vez que se ha identificado la causa, se definen las acciones correctivas apropiadas para evitar que vuelva a ocurrir. Una vez que estas medidas hayan demostrado ser efectivas y sostenibles, este proceso estará completo.
En principio, lo mismo se aplica a las acciones preventivas. Solo cuando se ha comprobado su eficacia se realiza un análisis de tendencias y todo se registra en el informe de gestión de calidad. Un enfoque tan rígido y pragmático no solo suena factible, sino que también es extremadamente práctico.
Hay varias razones por las que los sistemas CAPA, sin embargo, se han descarrilado con bastante facilidad en algún momento o todavía están en marcha. Por ejemplo, como suele ser el caso cuando se introduce un nuevo paradigma en la garantía de calidad farmacéutica, se observó un enorme aumento en las medidas CAPA en el período inicial cuando se establecieron los sistemas CAPA.
Esto se debió sólo en parte a una mayor conciencia de las interrelaciones pertinentes. Mucho más frecuentemente, se debió simplemente a malentendidos, como la evaluación de que toda no conformidad debe necesariamente resultar en una medida CAPA. Sin embargo, el Capítulo 1 de la Guía GMP de la UE no estipula la investigación de desviaciones o el inicio de medidas CAPA para cada no conformidad, sino que las limita a las desviaciones “relevantes”. Por lo tanto, es importante brindar a los empleados la capacitación suficiente y utilizar ejemplos para aumentar su conciencia sobre qué es una desviación y cómo debe documentarse.
Si una desviación documentada en el sitio se procesa más en el sistema CAPA o se puede corregir rápidamente en el sitio, lo decide una unidad superior o el control de calidad. En todo caso, deberá estar regulado en el SOP asociado.
Los involucrados en el proceso definitivamente deben participar en la evaluación de las no conformidades, porque conocen los procesos y las interrelaciones, tienen experiencia con las posibles causas y deben poder tomar decisiones rápidas.
La evaluación o clasificación rápida de un evento en particular no siempre tiene que ser realizada por un departamento de calidad superior. El experto en el sitio está, al menos según el entendimiento europeo de GMP, igualmente autorizado para ayudar a tomar decisiones. Como suele ser el caso, las mejores decisiones las toman todas las partes involucradas.
Es deseable, pero hasta ahora solo realizado en unas pocas empresas, vincular el instrumento de acción preventiva prospectiva con otros métodos de mejora continua de procesos como Six Sigma o Excelencia Operacional. Entonces, en lugar de usarse de forma rutinaria para cada desviación, en realidad solo podría aplicarse específicamente y con alta prioridad después de la evaluación de riesgos.
3 consejos para la vida diaria de GMP
Haga la vida un poco más fácil para los tomadores de decisiones y agregue una selección de medidas CAPA fijas al final del SOP para manejar las desviaciones, como cambios en las instrucciones de trabajo, cambios en los procesos, aumento en las frecuencias de prueba o monitoreo, cambio en las responsabilidades, o reentrenamiento de los empleados.
Use la medida estándar de “reentrenamiento de empleados” de uso frecuente con moderación. Una instrucción breve y directa a los empleados como medida correctiva suele ser más eficaz que una nueva formación formal.
Si la misma desviación o una similar ocurre una y otra vez, es hora de mirar más de cerca. Haga correcciones reales, tómese el tiempo para reescribir un SOP confuso y desordenado. Procesos, procedimientos, pautas y documentación más simples a menudo traen mejoras duraderas.
Este artículo se basa en el conocimiento de GMP contenido en el portal en línea GMP Compliance Adviser, que brinda información detallada sobre las mejores prácticas y regulaciones de GMP de Europa, EE. UU., Japón y muchos más (PIC/S, ICH, OMS, etc.). ).
Sobre el Autor:
Thomas Peither es miembro de la junta y director de marketing y desarrollo empresarial de GMP-Verlag Peither AG, Schopfheim, Alemania. Desde 2008, también dirige GMP Publishing Peither, Inc. Tiene más de 20 años de experiencia como consultor de GMP para empresas farmacéuticas. Se le puede contactar en thomas.peither@gmp-verlag.de.
Durante su visita a un laboratorio contratado de EE. UU., los inspectores de la FDA encontraron violaciones básicas de GMP en las siguientes áreas:
Manejo de resultados OOS (21 CFR 211.192).
Falta de verificación de los métodos de prueba (21 CFR 211.165(e))
Falta de integridad de los datos electrónicos (21 CFR 211.68(b))
Una mirada más cercana a las deficiencias descritas arrojará luz sobre por qué violan las reglas básicas de GMP en el laboratorio y por qué las respuestas de la empresa se consideraron inadecuadas, lo que finalmente condujo al envío de esta WL.
Manejo de resultados OOS
Después de que una prueba analítica arrojara resultados OOS, las mediciones se llevaron a cabo con una nueva preparación de muestra. De acuerdo con los procedimientos compatibles con GMP, primero se debería haber llevado a cabo una investigación de causa raíz para aclarar el motivo de los resultados OOS. Al revisar los documentos, los inspectores de la FDA también notaron que el SOP para el manejo de los resultados de OOS no contenía ninguna regulación correspondiente y obviamente le dio libertad al personal del laboratorio para proceder con tales casos, lo cual no es compatible con las reglas de GMP.
Los requisitos para manejar los resultados OOS se describen en la Guía para la investigación de resultados de pruebas fuera de especificación (OOS) de la industria para la producción farmacéutica.
La respuesta del laboratorio contratado a esta carta de deficiencia se limitó a afirmar que el SOP se había actualizado en consecuencia. Sin embargo, la FDA consideró inadecuada esta respuesta debido a que aún faltan regulaciones sobre la invalidación de los resultados de OOS. Además, no se realizó una revisión retrospectiva que pudiera haber servido como evidencia para identificar e investigar completamente todos los resultados de OOS.
Falta de verificación de los métodos de prueba
El laboratorio siempre utilizó el mismo método analítico para la determinación del contenido de la sustancia activa en productos terminados con diferentes formulaciones, sin proporcionar evidencia en cada caso de que el método también era adecuado para este propósito. De la misma manera, es decir, sin prueba de idoneidad, también se utilizaron los mismos métodos de la farmacopea para probar microorganismos en diferentes productos.
La FDA consideró que los datos obtenidos con los métodos de prueba no estaban libres de errores (precisos) y calificó como inadecuado el marco de tiempo dado en la respuesta para la realización posterior de los estudios de verificación del método.
Falta de integridad de los datos electrónicos
Los inspectores de la FDA encontraron que no se cumplían dos requisitos fundamentales de GMP relacionados con el manejo de datos electrónicos:
Control de acceso a los datos electrónicos
Audit Trail
El acceso a estos datos no estaba adecuadamente protegido, es decir, era posible eliminar registros. Además, no había rastro de auditoría en el software de procesamiento de datos de HPLC y cromatografía de gases y, por lo tanto, no había control sobre la posible manipulación de datos.
En sus comentarios a la FDA, la empresa aseguró que se había contactado a los fabricantes de los sistemas de cromatografía para actualizar el software en consecuencia. Sin embargo, dado que no se llevó a cabo una evaluación de estos sistemas con respecto a posibles eliminaciones de datos y riesgos potenciales para la calidad de los productos finales, la FDA también calificó estos comentarios como “inadecuados”.
Después de la descripción de las violaciones de GMP, la carta de advertencia contiene una breve sección sobre la opinión de la FDA sobre las responsabilidades de un laboratorio contratado. La declaración central de este pasaje es:
La FDA considera que el contratista es una extensión del sitio de producción del cliente. Las deficiencias de GMP en el contratista, por lo tanto, en principio, suponen un riesgo para la calidad de los medicamentos del cliente. De ello, la FDA deriva la obligación urgente de que el contratista informe a sus clientes de forma inmediata y completa sobre problemas con las analíticas.
El PIC/S (Pharmaceutical Inspection Co-operation Scheme) ya publicó una nueva versión de su Guía GMP PE016 en febrero. Esto se debe a la revisión del Reglamento de la UE nº 536/2014 sobre ensayos clínicos. El Anexo 13 del documento PIC/S ahora también se ha adaptado a esto. Esto está en línea con el acuerdo de cooperación entre PIC/S y EMA, que estipula que las guías PIC/S y EU GMP deben estar alineadas.
El Anexo 16 de la Guía PIC/S es nuevo. Describe la certificación por parte de la Persona Calificada y la liberación del lote. El Anexo 16 análogo de la Guía GMP de la UE no se incluyó en el documento PIC/S en 2016 porque el PIC/S consideró que el Anexo 16 era demasiado específico de la UE, especialmente porque la Guía PIC/S GMP se limita a la fabricación de medicamentos y no a la importación y distribución. Sin embargo, luego de una consulta con las autoridades del PIC/S en 2017, se acordó intentar implementar el Anexo 16 de la UE. El PIC/S también acordó que el elemento del Anexo 16 relacionado con los medicamentos importados es voluntario y depende de la legislación nacional.
La Guía GMP revisada (PE 009-16) con el Anexo 13 revisado y el nuevo Anexo 16 ya están vigentes y disponibles en el sitio PIC/S.
“Innovación superando la adversidad”. Esto se encuentra en el título de una publicación de la Dirección Europea de Calidad de Medicamentos y Cuidado de la Salud (EDQM) sobre el tema de las inspecciones remotas de GMP en tiempo real en los fabricantes de ingredientes activos (API) durante la pandemia de Covid-19.
Cuando se redactaron e introdujeron los requisitos y regulaciones de las BPM, nadie había pensado en una pandemia ni nada por el estilo. Luego, en la primavera de 2020, muchas cosas cambiaron. Además, EDQM, que supervisa el cumplimiento de las buenas prácticas de fabricación (GMP) y las solicitudes de certificados de idoneidad para las monografías de la farmacopea europea (CEP) en los sitios de fabricación de API, tuvo que reducir y adaptar su programa de inspección. Para ello, se concibió e implementó el concepto de “Inspecciones Remotas en Tiempo Real” (RTEMIS).
“Ninguna evaluación de BPF a distancia puede ser tan eficaz como una inspección in situ”; la EDQM fue consciente de esto desde el principio. Pero debería encontrarse una posibilidad que logre un “mayor grado de información sobre el cumplimiento de las BPF en comparación con el procedimiento de evaluación a distancia existente realizado exclusivamente sobre la base de documentos”. Por tanto, el objetivo era utilizar conexiones de vídeo en directo y en tiempo real entre los inspectores y las instalaciones a inspeccionar. Al hacerlo, debían tenerse en cuenta los siguientes desafíos técnicos, entre otros:
Seguridad de datos
Plataformas adecuadas para compartir documentos
Aplicaciones de conferencias web seguras
Ancho de banda sobre Wi-Fi o redes móviles (tasa de transferencia de datos de más de 100 kilobytes / segundo)
Diferentes zonas horarias
En septiembre de 2020, las cosas se pusieron en marcha y la EDQM se puso en contacto con los sitios de producción para una fase piloto voluntaria que ya había sido inspeccionada por la EDQM y tenía un buen historial de cumplimiento de las GMP.
Rápidamente se hizo evidente que este tipo de inspección remota requiere una preparación más minuciosa que las inspecciones in situ. También son importantes las conferencias telefónicas preparatorias, las pruebas de conectividad y una estrategia de redundancia.
El intercambio seguro de documentos confidenciales en tiempo real requería algo de reflexión. Para ello, EDQM utiliza su propia herramienta para compartir documentos. Otro problema técnico importante fue qué tipo de dispositivos móviles se podrían usar para capturar imágenes en vivo de áreas de fabricación potencialmente peligrosas. Sin embargo, ya existen teléfonos móviles “intrínsecamente seguros” para este propósito, que se han utilizado durante algún tiempo en otras áreas potencialmente explosivas como las refinerías.
El EDQM está satisfecho con los resultados del estudio. Aunque los inspectores no estaban en el sitio, se identificaron varias deficiencias menores y mayores.
¿A dónde vamos desde aquí?
Basadas en los principios de la gestión de riesgos de calidad, las inspecciones remotas se convertirán en parte del programa de inspección de EDQM. Éstos entran en juego principalmente en el caso de restricciones de viaje o si no se puede garantizar la seguridad de los inspectores. Las inspecciones remotas de GMP en tiempo real también deberían ser posibles con un historial de cumplimiento de GMP correspondiente de la instalación. Sin embargo, las inspecciones remotas no se utilizarán para evaluar los procesos de fabricación asépticos.
Las inspecciones remotas en tiempo real brindan al EDQM otra opción para evaluar el cumplimiento de GMP de un sitio. Al mismo tiempo, sin embargo, se enfatiza que estas inspecciones remotas no pueden reemplazar las inspecciones in situ en términos de valor y efectividad.
Fuente: sitio web de EDQM – News letter ECA, 14/7/2021
Los medicamentos están sujetos a regulaciones especiales. Es de vital importancia que los medicamentos no solo se fabriquen con una alta calidad de acuerdo con las Buenas Prácticas de Fabricación (GMP), sino que la calidad y la integridad de estos productos se mantengan a lo largo de toda la cadena de suministro al paciente. Aquí es donde entra en juego las Buenas Prácticas de Distribución (BPD). Las BPD es la parte del aseguramiento de la calidad que asegura que la calidad de los medicamentos se mantenga en todas las etapas de la cadena de suministro.
Distribución de productos farmacéuticos sensibles a la temperatura
En este momento, las vacunas COVID-19 en particular ilustran la importancia de la distribución de medicamentos sensibles a la temperatura y los desafíos involucrados. Por ejemplo, ¿cómo se debe transportar esta vacuna, pero también cualquier otro producto sensible a la temperatura en general, para llegar de manera segura desde los sitios de producción a los centros de almacenamiento y distribución y luego a los centros de vacunación locales?
En las Directrices para las BPD de la UE (Directrices de 5 de noviembre de 2013 sobre buenas prácticas de distribución de medicamentos para uso humano – 2013 / C 343/01), los requisitos especiales para productos sensibles a la temperatura se mencionan en el Capítulo 2.4. y Capítulo 9.4 .:
Capítulo 2.4 (Capacitación): „[…] El personal que se ocupe de cualquier producto que requiera condiciones de manipulación más estrictas debe recibir capacitación específica. Ejemplos de tales productos incluyen […] productos sensibles a la temperatura.
Debería mantenerse un registro de toda la formación, y la eficacia de la formación debería evaluarse y documentarse periódicamente “.
Capítulo 9.4. (Productos que requieren condiciones especiales): „[…] Para productos sensibles a la temperatura, se debe utilizar equipo calificado (por ejemplo, embalaje térmico, contenedores con temperatura controlada o vehículos con temperatura controlada) para garantizar que se mantengan las condiciones de transporte correctas entre el fabricante y el distribuidor mayorista. y cliente.
Si se utilizan vehículos con temperatura controlada, el equipo de control de la temperatura utilizado durante el transporte debe mantenerse y calibrarse a intervalos regulares. Se debe realizar un mapeo de temperatura en condiciones representativas y se deben tener en cuenta las variaciones estacionales.
Si se solicita, los clientes deben recibir información para demostrar que los productos cumplen con las condiciones de temperatura de almacenamiento. […]
El proceso para la entrega de productos sensibles y el control de las variaciones de temperatura estacionales deben describirse en un procedimiento escrito “.
Implementación
Es responsabilidad de la alta dirección en conjunto con la Persona Responsable (PR) asegurar que se implemente la formación inicial y continua del personal. La formación inicial debería incluirse en un procedimiento y describir claramente los requisitos mínimos y las tareas que, en consecuencia, se pueden realizar. También debe incluir detalles del proceso de aprobación.
Si el equipo utilizado no está calificado, los productos pueden verse afectados negativamente durante el transporte. Las empresas deben considerar las opciones disponibles, como vehículos con temperatura controlada y soluciones de embalaje activas o pasivas. Al tener un proceso documentado que tenga en cuenta las variaciones estacionales al definir los métodos de envío que se utilizarán para productos sensibles a la temperatura, se minimizará el riesgo de variaciones de temperatura.
El Subcomité Conjunto de Empaque y Distribución de la USP publicó una propuesta para el desarrollo de un nuevo Capítulo General <1xxx> Calificaciones de Proveedores. El Subcomité Conjunto hará recomendaciones al Comité de Expertos en Empaque y Distribución de los Capítulos Generales (CG), que será responsable de esta nueva norma. La fecha límite de entrada es el 25 de abril de 2021. Se espera que la propuesta final se publique para comentarios en el Pharmacopeial Forum (PF) 47 (5) [septiembre / octubre de 2021].
Calificación de proveedores basada en la gestión de riesgos y acuerdos técnicos.
Las diversas fuentes de material de empaque incluyen proveedores externos, locales y globales. Los proveedores externos con frecuencia también brindan servicios adicionales como ensamblaje, análisis, empaque, almacenamiento, transporte, distribución, etc. Esto significa que los componentes de empaque utilizados en el producto final deben fabricarse de acuerdo con las normas apropiadas (GMP). También significa que los procesos en la cadena de suministro de un material o servicio deben cumplir con las regulaciones relevantes y los requisitos (GDP-) (si corresponde).
Según la USP, los fabricantes de productos farmacéuticos deben tener procesos para evaluar sistemáticamente a los proveedores de envases sobre:
El riesgo para la calidad del material o servicio suministrado,
Cumplimiento de los sistemas de calidad con las regulaciones relevantes y los requisitos (GMP / GDP-), si corresponde,
La confiabilidad del proveedor para evitar desviaciones de calidad y escasez de componentes y / o materiales de empaque,
Un contrato de suministro o acuerdo de calidad.
El enfoque incluye la identificación y selección, evaluación y aceptación, monitoreo del desempeño y descalificación de proveedores de materiales de empaque (por ejemplo, empaque primario, secundario, auxiliar y de envío) y proveedores de servicios (por ejemplo, proveedores de logística para almacenamiento y transporte, servicios analíticos).
Muchos de los requisitos establecidos en las regulaciones GMP de los Estados Unidos de NA (21 CFR 210/211) no son muy específicos. Sin embargo, se concretan, entre otras cosas, en las Guías y en las Cartas de Advertencias o Warning Letters (WL). Estas últimas en particular muestran muy claramente cómo la FDA interpreta sus regulaciones GMP. Una WL actualizada proporciona información sobre las tareas de la Unidad de Garantía de Calidad.
En una WL actual con fecha del 22 de enero de 2021, la FDA criticó las deficiencias en la Unidad de Garantía de Calidad de un fabricante chino de medicamentos de venta libre (OTC).
En particular, la FDA criticó la falta de procedimientos escritos sobre las responsabilidades de la Unidad de Calidad con respecto a:
Revisión de registros por lotes (BRR)
Calificación de proveedores
Calificación de equipos
Calibración
Validación de procesos
Validación de limpieza
Revisión anual de productos (APR)
Un plan de acción para corregir las deficiencias debe demostrar que las instrucciones de procedimiento en la empresa son adecuadas y “sólidas”. Se deben tomar medidas para garantizar que la Unidad de Garantía de Calidad pueda aplicar su “Supervisión de la Calidad” de manera amplia. En particular, las investigaciones y, si es necesario, la clasificación de las mercaderías con respecto a su identidad, calidad y pureza se abordan como tareas de la Unidad de Garantía de Calidad.
Además, la FDA criticó que no se hayan realizado pruebas sobre los materiales de partida y los productos finales con respecto a la identidad y el contenido. También se quejó de la falta de datos de estabilidad que confirmen la vida útil de los productos.
Todo esto resultó en una clara referencia a la Guía de la FDA “Enfoque del sistema de calidad para las regulaciones farmacéuticas cGMP”. La FDA también recomienda contratar a un consultor, teniendo en cuenta los requisitos de 21 CFR 211.34. Este consultor primero debe realizar una auditoría de todas las actividades relevantes de GMP y verificar la implementación y efectividad de las medidas CAPA basadas en la WL. Solo después de eso, la compañía debe comunicarse nuevamente con la FDA. La WL enfatiza expresamente que la Alta Dirección es responsable de remediar las deficiencias.
Conclusión: En esta WL, la FDA proporciona información muy clara sobre las tareas integrales de una Unidad de Garantía de Calidad, incluida una “Supervisión de la Calidad”, de acuerdo con las regulaciones de GMP de EE. UU.
La ICH Q9 (Gestión de Riesgos de Calidad), es la principal guía que proporciona principios y ejemplos de herramientas para la Gestión de riesgos de calidad (GRC) que se pueden aplicar a diferentes aspectos de la calidad farmacéutica.
La GRC en sí misma desempeña un papel importante en las directrices de las cGMP. De hecho la nueva GMP de la ANMAT (Disposición 3827/2018) tiene un anexo, el número 8, dedicado a este tema.
Las cGMP indican que para alcanzar el objetivo de calidad de manera confiable, debe existir un sistema de Garantía de Calidad integrado y correctamente diseñado que incorpore buenas prácticas de fabricación, control de calidad y gestión de riesgos de calidad. Debería estar completamente documentado y su efectividad monitoreada.
Algunos ejemplos de áreas GMP donde se usan los principios de gestión de riesgos son:
Gestión de desviación y CAPA
Control de cambios
Determinación del alcance y alcance de las actividades de calificación y validación
Procesos de monitoreo y muestreo
Revisiones de documentos
Calificación del proveedor
Gestión de materiales
La ICH Q9 describe algunas de las herramientas más importantes para implementar los respectivos principios de gestión y evaluación de riesgos. Las actividades de gestión de riesgo de calidad usualmente son llevadas a cabo por equipos interdisciplinarios, incluyendo a los expertos de las áreas apropiadas.
Pero, ¿cómo observan las autoridades competentes estos sistemas en sus inspecciones de GMP?
Si el proceso de gestión de riesgos de calidad, se basa en la ICH Q9, probablemente se apoyarán en dicha guía, como referencia, buscando ver como es la relación de los sistemas e calidad con la GRC, si el concepto de riesgo está claro, la organización de gestión de riesgo de calidad, sus responsables y la agenda de actividades. Decisiones y seguimiento de acciones definidas a través de la GRC. Cómo está relacionada la GRC con la mejora continua.
Como ejemplo de algunas de las observaciones identificadas, podemos mencionar, no disponer de análisis de riesgos de los cambios que se efectúan en la planta, o planes de auditorías a proveedores no están basados en un análisis de riesgos de los mismos.
Espero que les resulte útil y que si tienen un proceso de GRC lo revisen y en el caso que aún no lo están haciendo, empiecen a implementar este proceso, que tiene un enfoque netamente preventivo y está orientado a la mejora continua.