En Julio 2022, fue publicada la segunda edición de la Guía ISPE GAMP® 5: un enfoque basado en el riesgo para sistemas computarizados compatibles con GxP.
Esta segunda edición de ISPE GAMP 5 resalta la importancia de los proveedores de servicios, del mayor uso de herramientas de software y automatización para lograr un mayor control, una mayor calidad y menores riesgos a lo largo del ciclo de vida y además destaca la importancia del “pensamiento crítico”.
En este punto quiero detenerme y dejarles un resumen de un artículo escrito por Charlie C. Wakeham, sobre Por qué necesitamos el pensamiento crítico:
Puede parecer que las únicas cosas que la industria farmacéutica ama más que los acrónimos son las nuevas frases de moda. Y eso puede ser verdad. Pero a veces, detrás de la frase de moda, hay beneficios reales, fuertes y alcanzables para nuestro trabajo, nuestra industria y nuestros pacientes finales.
El pensamiento crítico es una de esas frases de moda. Originadas fuera de nuestra industria y adoptadas con entusiasmo por instituciones académicas y consultores a nivel mundial, la mayoría de las definiciones son algo ininteligibles y difíciles de relacionar con la vida real.
Charlie menciona que hay no menos de 11 definiciones, pero me voy a quedar con la que para él es su propia definición:
Es elegir sabiamente, excluyendo la burocracia, la jerarquía, el ego, el sesgo y la ambición del proceso de toma de decisiones.
Se está enfocando en lo que importa, que tiene que ser la calidad en lugar del cumplimiento y la documentación. Obtenga la calidad correcta y el cumplimiento estará allí de todos modos.
Me parece más importante lograr que la industria utilice el pensamiento crítico que debatir las complejidades del paradigma.
Veamos algunos ejemplos de pensamiento crítico con sistemas computarizados GxP:
ESPECIFICACIONES DE REQUISITOS
Todos estamos de acuerdo en que necesitamos capturar lo que debe hacer un sistema computarizado cuando estamos planeando un nuevo sistema, es decir, la funcionalidad que proporcionará para respaldar un proceso de negocios en particular. Esto significa que debemos definir qué regulaciones se aplican y qué requisitos individuales deben cumplirse dentro del marco regulatorio general. Necesitamos requisitos para definir la funcionalidad específica que el sistema debe proporcionar en relación con el proceso comercial; cómo controlará una actividad, analizará una muestra, calculará un resultado y almacenará los datos. Puede haber restricciones particulares o requisitos de infraestructura: alojamiento en la nube, funcionamiento en una sala limpia, conexión o interfaz con un sistema existente.
Todos estos son tremendamente importantes y deben ser capturados como requisitos.
Requisitos sobre cuántos niveles de submenú se permiten en el sistema. Requisitos para “rápido, fácil, fácil de usar”. Estos no tienen sentido para el proceso comercial e inútiles para el paciente final.
¿Qué hay de capturar los requisitos en un documento de Word en lugar de utilizar una herramienta de gestión de requisitos? ¿Qué elección de logotipo, fuente, interlineado? ¿Serán los requisitos firmados a mano o aprobados electrónicamente? Nada de esto tiene ningún impacto en la calidad del producto o la seguridad del paciente. Siempre que los requisitos estén controlados, aprobados, protegidos contra cambios no autorizados, disponibles y actualizados durante toda la vida del sistema, el formato, los medios y la apariencia de la información tampoco tienen impacto en la integridad de los datos.
Mencioné “aprobado” en la lista anterior. Revisión por el propietario del proceso y el propietario del sistema, absolutamente. Revisión por otras cuatro personas sin conocimiento de sistemas o procesos que solo querían su nombre en el documento, sin sentido. Mi registro personal en un proyecto CSV era un cliente que demandaba un total de nueve revisores y aprobadores. Si un revisor no va a leer el contenido o entender el contenido, ¿qué representa su firma: un autógrafo para la posteridad? ¿Una confirmación de que creen que el crítico anterior probablemente lo leyó? No ayuda….
PRUEBAS
Se pueden obtener grandes beneficios al aplicar el pensamiento crítico a las pruebas y, por lo tanto, no sorprende que la excelente instigación de la FDA de los enfoques de Computer Software Assurance (CSA) tenga un enfoque significativo en las pruebas.
Yo estimaría de forma conservadora que los scripts de prueba detallados click a click tardan cinco veces más en escribirse que en ejecutarse. La mayor proporción de defectos encontrados por dichos scripts de prueba son, sin duda, errores en los propios scripts. Veamos la motivación para scripts de prueba tan detallados.
Motivación Pensamiento Crítico
Debe desafiar una ruta o rama específica en el software; las instrucciones detalladas aseguran que llegue al camino o ramal. Esto aporta valor en la detección de defectos y beneficios finales para la seguridad del paciente.
El evaluador no sabe cómo operar el software, por lo que necesita las instrucciones de click a click.
Posts tagged ‘ISPE’
La validación de limpieza es una actividad requerida dentro de la Industria Farmacéutica.
Desde las regulaciones y los estándares de la Industria, la validación de limpieza es reconocida como una actividad de suma importancia para establecer que la contaminación cruzada de un producto está controlada de manera de asegurar la calidad del producto y la seguridad del paciente.
Es una actividad continua dentro de las cGMP, la cual necesita una inversión significativa de recursos y de tiempo.
Si tomamos como una referencia la guía ISPE, Validación de limpieza para el siglo 21, uno de los objetivos que desarrolla es la aplicación del análisis de riesgo basado en la ciencia, en el proceso de validación de limpieza.
Análisis de riesgo de la limpieza
Riesgo de acuerdo a los principios de la ICHQ9 es función de la severidad del peligro y la probabilidad de ocurrencia.
Para los propósitos de la validación de limpieza, podemos definirlo como:
Riesgo = Fc (Severidad del residuo, Prob. aparición del residuo, Detectabilidad del residuo)
El nivel de esfuerzo, formalidad y documentación del proceso de QRM (Quality Risk Mangement) debe ser proporcional al nivel de riesgo del proceso de limpieza.
Análisis de Riesgo de la limpieza
El objetivo de la inspección es asegurar que las bases para todo límite están justificadas científicamente, por lo tanto los límites deberían ser determinados a partir del peligro que el residuo del proceso representa. Esto puede ser determinado desde la revisión de la toxicidad de los mismos residuos.
El ADE (límite de exposición diaria aceptable), puede ser usado como una medida de severidad del peligro de la sustancia, y a partir del mismo efectuamos el cálculo del MSC (Máxima cantidad arrastrada permitida segura).
Además debe ser considerado el peligro potencial aportado por el diseño del equipo, por ejemplo considerar que el equipo sea fácil de limpiar, inspeccionar y muestrear.
En cuanto a los agentes de limpieza, los mismos deben ser seleccionados en base a principios científicos y con conocimiento de los peligros que pueden representar. Es deseable que los mismos estén incluidos dentro de la lista GRAS (Generally Recognized As Safe), en caso que no se encuentren dentro de esta lista, puede ser usado el ADE del mismo para determinar su nivel de riesgo.
También se debe evaluar el peligro de una posible carga biológica remanente y la posibilidad de proliferación microbiológica durante o después de un proceso de limpieza y los peligros que esto representa. Por ejemplo, es necesario abordar los peligros que se presentan al mantener el equipo en estado sucio o limpio.
Exposición de la limpieza
Luego que el peligro de una sustancia ha sido identificado, a partir de su ADE se determina la MSC (máxima cantidad permitida segura), siguiendo las etapas para minimizar y evaluar los niveles posibles de exposición.
Antes de usar un SOP de limpieza, debe ser efectuado un análisis de riesgos (RA) por ejemplo utilizando FMEA. Se calcula el RPN (Risk Priority Number), a partir de la severidad del residuo, el nivel del residuo y la posibilidad de detectarlo. Cuando el RPN es alto, deben ser tomadas acciones de mitigación de forma de disminuir el riesgo.
Si el riesgo no puede ser reducido a nivel aceptable, el producto podría requerir equipos dedicados para su elaboración o el uso de elementos desechables.
Cuando el RA indica que la contaminación microbiológica es una preocupación, tales como equipamiento estéril, holding time de equipos, etc. Deben ser obtenidos y evaluados datos, para determinar el nivel de exposición. Caso contrario, los datos microbiológicos pueden no ser necesarios.
Detección de limpieza
La habilidad para detectar un residuo cuando está presente, es un factor importante en la reducción del riesgo.
Hay distintos métodos para la determinación de residuos, inspección visual, TOC, conductividad, HPLC, etc.
La inspección visual de determinados residuos, método usado para la verificación de la limpieza, apropiado para productos de bajo riesgo.
La conductividad, es otro método utilizado para la detección de presencia de productos de bajo riesgo. Es muy utilizada para la detección del punto final en los sistemas CIP (Clean In Place).
TOC, Carbono Orgánico Total, es una herramienta poderosa, simple y rápida, aunque inespecífica para la detección de residuos de limpieza.
HPLC, herramienta muy sensible, específica, para detectar residuos. Es de amplio uso para la detección de residuos de validación de limpieza. Es el método cuando los anteriores no pueden ser utilizados. usualmente se parte del método de valoración del activo y se lo adapta al nivel de trazas y luego se valida.
Debemos disponer de procedimientos de limpieza que nos aseguren procesos de limpieza de alta Capacidad y en el caso de sustancias de alta toxicidad, disponer de un monitoreo apropiado de los mismos.
- Requerimientos basados en el producto / proceso (pensamiento basado en el producto / proceso)
- Risk Assessment basado en la seguridad del paciente
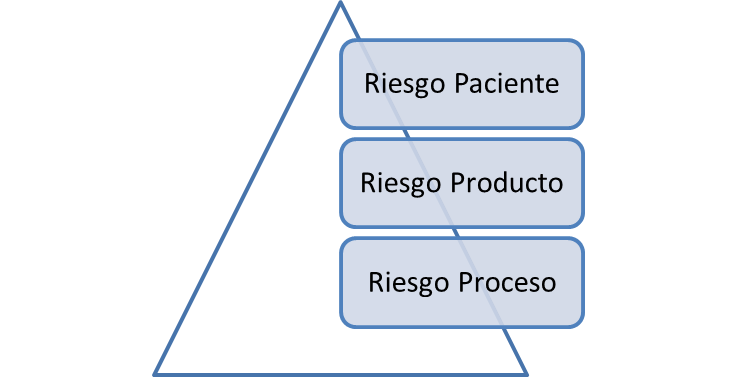
Cuando pensamos en Riesgo al producto, debemos considerar la seguridad, identidad, pureza, calidad y potencia del producto.
En cuanto a riesgos del proceso, debemos considerar los parámetros críticos del proceso, como temperaturas, tiempos de operación, presiones, IPCs, etc.
- Aumento del uso de las GEP (Buenas Prácticas de Ingeniería) – uso del FAT /SAT / Commissioning. Formas de trabajo de ingeniería.
- Testeos / ensayos de acuerdo al riesgo
- Reutilización de trabajos previos (ha sido hecho por otros, podemos confiar en otros) – know how, buenas prácticas
Según ISPE:
Requisitos: el URS debe basarse en el proceso y esto se confirma en el PQ (calificación de performance). Esto significa que tanto el IQ como el OQ están subordinados en importancia.
Durante bastante tiempo hemos tenido en mente que los procesos de validación y de calificación debían contemplar dentro de los procedimientos cuál era la frecuencia con que debíamos repetir la actividad, sin embargo las cGMP nos van orientando hacia implementar una serie de actividades sistemáticas que nos permitan revisar y documentar que nuestros equipos, procesos, sistemas, etc. mantienen el estado validado o calificado. Por ejemplo en sistemas computarizados hablamos de efectuar una revisión periódica de los sistemas computarizados validados, en el caso de los procesos validados hablamos de seguirlos a través de la verificación continua del proceso o de la Revisión Anual de Productos.
¿Cuáles son los ciclos adecuados para la recalificación de equipos?
Con la revisión del Anexo 15 en octubre de 2015, el tema de la recalificación se ha vuelto más importante. En el antiguo Anexo 15 de 2001, el tema de la recalificación estaba “oculto” bajo la demanda general de revalidación (punto 45). Con la revisión del Anexo 15, la recalificación ahora tiene su propio capítulo con los requisitos para:
- Una evaluación del equipo con la frecuencia adecuada para demostrar que se ha mantenido en un estado de control y
- Cuando es requerida una recalificación, los intervalos de tiempo deben justificarse y los criterios para la evaluación deben establecerse.
En la práctica, a veces es difícil establecer tales programas y criterios para la evaluación. En el área estéril, a veces hay referencias específicas de las regulaciones a dispositivos y procesos individuales. Por ejemplo, de acuerdo con la Guía aséptica de la FDA, los filtros HEPA en sala limpia clase 5 deben probarse dos veces al año. El Anexo 1 de las Directrices GMP de la UE también proporciona directrices sobre este tema (por ejemplo, para la esterilización).
¿Qué pasa con el equipo en otras áreas?
En este caso, puede ser útil el capítulo 9 de la línea de base ISPE n° 5 revisada Puesta en servicio y calificación de junio de 2019 sobre la revisión periódica. El enfoque de “revisión”, es decir, la evaluación, se presenta en dos fases.
En la fase 1, se clasifican los “sistemas de impacto directo” existentes. Dependiendo de la complejidad del sistema (complejo vs. estándar) y la influencia en la calidad del producto, los sistemas se dividen de manera ejemplar en las categorías 0-3. Cada categoría, excepto la categoría 0, se asigna a un período de “revisión”. La categoría 0 se refiere a los sistemas existentes donde tenemos disponibles datos de monitoreo. En este caso, no es necesaria una revisión de estos sistemas de categoría 0 (por ejemplo, sistemas de agua). Ya que tenemos datos para su evaluación. Para sistemas en la categoría 1, por ej. autoclaves, se aplican las especificaciones anteriores de las reglamentaciones estériles. Para sistemas de categoría 2, por ej. tanques de almacenamiento intermedio, la línea de base sugiere un intervalo de dos años. Para sistemas de categoría 3, por ej. comprimidoras, la línea de base recomienda un intervalo de revisión de tres años.
En la fase 2, se ejecuta la “revisión”. La revisión en sí tiene lugar como un proceso de tres pasos:
Paso 1: una evaluación inicial con respecto al cumplimiento de GMP, historial de cambios, mantenimiento / calibración y desviaciones.
Si esta evaluación plantea dudas sobre el impacto del sistema en la calidad del producto, se debe seguir el paso 2. De lo contrario, el proceso termina aquí.
Paso 2: en función del resultado del paso 1, los expertos en la materia (SME) examinan en mayor profundidad los sistemas examinados en el paso 1, cuyos resultados son cuestionables. Los SMEs deben provenir de los mismos departamentos que también participaron en la evaluación en el paso 1.
Si la evaluación en el paso 2 llega a la conclusión de que un sistema ya no puede estar en estado calificado, el siguiente paso, el paso 3, tiene lugar. De lo contrario, el proceso termina aquí.
Paso 3: en función del resultado del paso 2, los SMEs examinan en mayor profundidad los sistemas examinados en el paso 2, cuyos resultados son cuestionables. Los SMEs deben provenir de los mismos departamentos que también participaron en la evaluación en los pasos 1 y 2. Si es necesario, se deben tomar medidas para que el sistema vuelva a un estado calificado. Se debe crear un documento de desviación para controlar las acciones tomadas.
De acuerdo con la línea de base, todo el proceso debe ser administrado por alguien con una gran experiencia. La revisión en sí misma debe al menos ser aprobada por el propietario del sistema y la unidad de calidad. Un formulario de revisión es parte de la línea de base como Apéndice 11 y se da un ejemplo en el Apéndice 12.
Tomado de la news Letter de la ECA (28/8/2019)
Si miramos las observaciones de las agencias como la FDA y la EMA, la validación de procesos y la calificación de equipos están dentro del top ten de deficiencias encontradas. Además de deficiencias relacionadas con el diseño de locales y equipos, y pobres instrucciones sobre calibración, limpieza y mantenimiento de los equipos.
¿Cómo podemos llevar a cabo una calificación que cumpla con las cGMP y, además sea eficiente?
Matthias Klein de CSL-Behring, reconoce que la clave del éxito radica en el uso eficiente de los documentos y procesos existentes (incluidos los sistemas de control de cambio y calibración) en las compañías farmacéuticas. La minimización del gasto también es posible aplicando de manera consistente las GPE de acuerdo con la Guía de referencia del ISPE sobre commissioning y calificación como un concepto de calificación integrado. Podemos mencionar los documentos de pruebas de aceptación de fábrica (FAT) o las pruebas de aceptación del sitio (SAT). Estas pruebas efectuadas por personal capacitado, y documentadas adecuadamente, nos permitirá evitar la duplicación de trabajo.
Para una efectuar una calificación eficiente y pragmática debemos usar el análisis de riesgos del proceso de manera apropiada (la ICH Q9 da los lineamientos y una serie de herramientas, entre las que mencionamos el FMEA).
El análisis de riesgos apropiado reduce aún más el esfuerzo de calificación.
Un análisis de riesgo general es la base para la calificación de diseño (DQ). Los componentes relevantes para la calificación son identificados y el grado de la calificación se determina mediante un análisis detallado del riesgo. El análisis de riesgo también debe considerarse durante el curso de la calificación y en la operación de rutina.
Durante el proyecto de calificación, el control de cambios se puede manejar de manera casi pragmática.
Las razones para las pruebas de calificación se cuestionan cada vez más en el contexto de las inspecciones y auditorías en las que el concepto integrador y una matriz de trazabilidad adecuada pueden garantizar que solo se realicen las pruebas que provienen del análisis de riesgos.
En cuanto a la recalificación ahora se requiere una evaluación periódica de los locales, sistemas, equipos y servicios en relación con el estado de calificación. También se deben evaluar pequeños cambios. La frecuencia de esta evaluación debe ser justificada.
La Guía de la FDA ya no contiene el término revalidación. Con el enfoque del ciclo de vida, el tema continúa en la etapa 3 Verificación continua del proceso, cuyo objetivo es mostrar que el proceso permanece en un estado validado durante la producción de rutina. Para este propósito, se solicita un sistema que detecte desviaciones de proceso no planificadas. El proceso no debe salirse de control. Aquí se hace una referencia directa a la Revisión Anual del Producto (21 CFR 211.180) para apoyar este programa en curso. Y se atribuye gran importancia a las estadísticas.
¿Y el futuro?
El ISPE y la ASTM (Sociedad Americana de Pruebas y Materiales) apuntan a una “calificación de valor agregado”. Debemos enfocarnos en las especificaciones de requisitos del usuario (URS) y la Calificación de proceso (PQ).
El ISPE resume los “Principios para la Calificación del Siglo 21” en diez puntos, que es un excelente resumen de los principios descritos en la norma ASTM E2500:
- Enfoque en la calidad del producto.
- “Requisitos del usuario” basados en el proceso. Se confirman como satisfechos en el PQ (IQ y OQ están subordinados en importancia).
- “Evaluaciones de riesgo”: el desarrollo del proceso y el diseño experimental son los elementos clave para identificar funciones y parámetros críticos.
- Solo se utilizarán los parámetros críticos del proceso como base para la calificación.
- Todas las actividades deben aportar valor al proceso (“No haremos nada solo por el cumplimiento de la normativa”).
- Actividades de calificación basadas en el riesgo. Ej. clasificación GAMP.
- “Documentos de valor agregado”.
- Uso de la documentación del proveedor, si es posible.
- Pruebas: por regla general, deben realizarse una sola vez. Pero puede ser necesario repetir algunas pruebas que se han llevado a cabo en una etapa anterior del desarrollo.
- Fomento de la innovación: se requiere la flexibilidad de los programas de calificación para poder implementar nuevas tendencias.
Todo esto requeriría grandes cambios organizativos en las empresas (probablemente uno de los mayores obstáculos) que a menudo tienen conceptos relativamente rígidos de calificación y validación.
El riesgo para la calidad del producto se deberá basar en el conocimiento del proceso.
Este proceso tiene lugar bajo el paraguas de las Buenas Prácticas de Ingeniería y sobre la base de la gestión de riesgos, la gestión de cambios y la revisión del diseño. El input al proceso es el conocimiento del producto, el conocimiento del proceso, los requisitos reglamentarios y los requisitos de calidad de la empresa.
El output esperado, es la operación y la mejora continua.
En la Guía de Validación de Procesos de la FDA, las actividades de calificación son parte de la Etapa 2 del ciclo de vida de validación del proceso de calificación del proceso. Los términos DQ, IQ y OQ ya no se usan en el documento. Esto es consistente con la Guía Estándar ASTM E2500 y GAMP 5. Ambos documentos se abstienen de usar estos términos y los sustituyen por “verificación”. La intención es dejar claro que las prácticas de la industria de DQ, IQ y OQ, etc. no son un requisito reglamentario y fomentar una aplicación más sólida de las Buenas Prácticas de Ingeniería (GEP).
La revisión del Anexo 15 va en la dirección de las GEP y el FAT y SAT, y si bien menciona etapas de calificación como DQ, IQ, OQ, PQ las mismas no son obligatorias.
De acuerdo a la Guía de Validación de Procesos de la FDA los lotes de la Calificación de Rendimiento del Proceso (PPQ), deben ser llevados a cabo por empleados de producción calificados utilizando instalaciones y equipos calificados; bajo las condiciones de fabricación comerciales y, en última instancia, pueden ser lanzados como lotes comerciales normales, dependiendo de un resultado de Calificación de Proceso global exitoso.
De acuerdo a la FDA “Una PPQ exitosa confirmará el diseño del proceso y demostrará que el proceso de fabricación comercial funciona como se esperaba”. Por lo tanto, la terminación de PQ antes de la comercialización es obligatoria.
La FDA considera PPQ en el sentido del documento PIC / S PI 006. Ha igualado PQ y la validación de procesos desde 1996. En el Anexo 15 revisado, PQ y Process Validation se pueden combinar.
Conclusión
El futuro de la calificación parece residir en una calificación aún más integrada que involucra a ingenieros y calificadores, en un proyecto estructurado basado en el análisis de riesgos e involucrando la mayor cantidad posible de documentos GEP. Las pruebas de los preceptos URS en el PQ serán la base para las principales pruebas de calificación. Seguramente el nuevo modelo de “verificación” de la norma ASTM E2500 reemplazará las etapas de calificación clásicas.
El futuro de la validación estará enfocado en la comprensión del proceso. Las estadísticas serán útiles para apoyar estos aspectos.
En Europa, el enfoque “tradicional” seguirá estando disponible como se indica en la Guía de la Validación de Procesos de la EMA y en el Anexo 15 revisado. Este enfoque tradicional ya no se menciona en la Guía de la FDA.
¿Qué hay de los productos heredados (productos antiguos)? La FDA dice que comienza con la etapa 3 “Verificación continua del proceso”. En el Anexo 15 revisado no hay pistas sobre productos heredados, aunque el enlace a PQR indica que los productos heredados también están en el foco de la verificación del proceso en curso. El enfoque de EMA es muy intensivo en la “verificación continua del proceso” ahora, más que en la Guía de la FDA.
Hasta ahora, a menudo se consideraba que la PQ estaba relacionada principalmente con el equipo. La perspectiva cambió en la guía de la FDA, con la nueva expresión PPQ como parte de PQ (ahora conocida como Calificación de rendimiento del proceso).
Tomado de: ECA Validation Good Practice Guide
Estos 5 conceptos claves son aplicados a lo largo de la GAMP5:
- Comprensión de productos y procesos
Una comprensión del proceso soportado es fundamental para determinar los requisitos del sistema. La comprensión de productos y procesos es la base para tomar decisiones basadas en la ciencia y el riesgo para garantizar que el sistema sea adecuado para el uso previsto.
Los esfuerzos para garantizar la aptitud para el uso previsto deben centrarse en aquellos aspectos que son críticos para la seguridad del paciente, la calidad del producto y la integridad de los datos. Estos aspectos críticos deben ser identificados, especificados y verificados.
Los sistemas dentro del alcance de la GAMP5 admiten una amplia gama de procesos, incluidos ensayos clínicos, estudios toxicológicos, producción de API, producción de productos formulados, almacenamiento, distribución y farmacovigilancia.
Para algunos sistemas de fabricación, los requisitos del proceso dependen de una comprensión profunda de las características del producto. Para estos sistemas, la identificación de Atributos de calidad críticos (CQA) y los Parámetros críticos de proceso (CPP) relacionados permiten la definición de los requisitos de control de proceso.
La especificación de requerimientos debe estar enfocada en aspectos críticos. El alcance y el detalle de la especificación de requisitos deben ser acordes con el riesgo, la complejidad y la novedad asociados del sistema.
La comprensión incompleta del proceso dificulta el cumplimiento efectivo y eficiente y el logro del beneficio comercial.
- Enfoque del ciclo de vida dentro de un QMS
Adoptar un ciclo de vida completo del sistema computarizado implica definir las actividades de una manera sistemática desde la concepción del sistema hasta su retiro. Esto permite el control de la gestión y un enfoque coherente en todos los sistemas.
El ciclo de vida debe formar parte intrínseca del QMS de la empresa, que debe mantenerse actualizado a medida que se desarrollan nuevas formas de trabajo.
A medida que se adquiere experiencia en el uso del sistema, el sistema de gestión de la calidad del servicio (QMS, por sus siglas en inglés) debe permitir mejoras continuas en el proceso y en el sistema basadas en revisiones y evaluaciones periódicas, datos operativos y de rendimiento, y análisis de causas de fallas. Las mejoras identificadas y las acciones correctivas deben seguir a la gestión del cambio.
Un ciclo de vida adecuado, aplicado adecuadamente, permite garantizar la calidad y la aptitud para el uso previsto, y lograr y mantener el cumplimiento de los requisitos reglamentarios. Un ciclo de vida bien gestionado y comprendido facilita la adopción de un enfoque QbD.
El enfoque del ciclo de vida es fundamental para la GAMP5 y representa cada uno de los otros conceptos claves.
- Actividades escalables del ciclo de vida
Las actividades del ciclo de vida se deben escalar de acuerdo con:
- Impacto del sistema en la seguridad del paciente, la calidad del producto y la integridad de los datos (evaluación de riesgos)
- Complejidad y novedad del sistema (arquitectura y categorización de los componentes del sistema).
- Resultado de la evaluación del proveedor (capacidad del proveedor)
El impacto sobre el negocio también puede influir en la escala de las actividades del ciclo de vida.
La estrategia debe estar claramente definida en un plan y seguir las políticas y procedimientos establecidos y aprobados.
- Gestión de riesgos de calidad basada en la ciencia
La gestión de riesgos de calidad es un proceso sistemático para la evaluación, control, comunicación y revisión de riesgos.
La aplicación de Quality Risk Management permite que el esfuerzo se centre en aspectos críticos de un sistema computarizado de manera controlada y justificada.
La gestión de riesgos de calidad debe basarse en una clara comprensión del proceso y un impacto potencial en la seguridad del paciente, la calidad del producto y la integridad de los datos. Para los sistemas que controlan o monitorean los CPP, estos deben ser rastreados a los CQA y, en última instancia, a las presentaciones reglamentarias relevantes para los sistemas de fabricación.
Se pueden utilizar técnicas cualitativas o cuantitativas para identificar y gestionar los riesgos. Los controles están desarrollados para reducir los riesgos a un nivel aceptable. Los controles implementados son monitoreados durante la operación para asegurar la efectividad continua.
- Aprovechamiento de la participación del proveedor
Las compañías reguladas deben tratar de maximizar la participación del proveedor a lo largo del ciclo de vida del sistema para aprovechar el conocimiento, la experiencia y la documentación, sujeto a una evaluación satisfactoria del proveedor.
Por ejemplo, el proveedor puede ayudar con la recopilación de requisitos, las evaluaciones de riesgos, la creación de especificaciones funcionales y de otro tipo, la configuración del sistema, las pruebas, el soporte y el mantenimiento.
La planificación debe determinar la mejor manera de utilizar la documentación del proveedor, incluida la documentación de prueba existente, para evitar duplicación de esfuerzos. La justificación para el uso de la documentación del proveedor debe ser proporcionada por el resultado satisfactorio de las evaluaciones del proveedor, que puede incluir auditorías del proveedor.
La documentación debe evaluarse para determinar su idoneidad, precisión y exhaustividad. Debe haber flexibilidad con respecto a las prácticas aceptables de formato, estructura y documentación.
ISPE GAMP 5 A Risk Based Approach to Compliant GxP Computarized Systems (2008).
El ISPE nos da algunos principios para efectuar las actividades de calificación:
- Enfoque en la calidad del producto: la calidad del producto debe ser garantizada por las actividades de calificación.
- Requisitos: el URS debe basarse en el proceso y esto se confirma en el PQ (calificación de performance). Esto significa que tanto el IQ como el OQ están subordinados en importancia.
- Evaluaciones de riesgo: el desarrollo del proceso y el diseño experimental son los elementos clave para identificar funciones y parámetros críticos, los cuales serán la base para las actividades de calificación.
- Actividades basadas en el riesgo, las mismas dependerán de la complejidad de la instalación (ej. Clasificación GAMP) y deben agregar valor a la calificación. No deberíamos incorporar solo actividades por el hecho de hacer volumen de documentación.
- Uso de la documentación del proveedor: Si es posible.
- Pruebas: por regla general, las pruebas deben realizarse una sola vez.
- Fomento de la innovación: en este punto, se requiere la flexibilidad de los programas de calificación para poder implementar nuevas tendencias.
Todo esto requiere grandes cambios organizativos en las empresas (quizás es una de las partes más difíciles), que a menudo tienen conceptos relativamente rígidos de calificación y validación.
La gestión de calidad se centraría más en los procesos y análisis de riesgo, los planes de calificación y los informes de calificación. Por otro lado, también sería necesario fortalecer los sistemas de calidad de los departamentos especiales (por ejemplo, Ingeniería, IT, etc.) y la producción. El riesgo para la calidad del producto se determinaría en el contexto de una divulgación de riesgo sobre la base de la comprensión del proceso. El control de esos riesgos se calificaría entonces en la calificación de la performance.
La nueva disposición de la ANMAT tiene establecidos requerimientos para los sistemas computarizados o informatizados en el Anexo 6 Sistemas Informatizados de la nueva Disposición.
Este Anexo aplica a todas las formas de sistemas informatizados usados como parte de las actividades reguladas por las BPF y se utilicen para crear, modificar, mantener, archivar, obtener o distribuir registros electrónicos.
Además de Validar las aplicaciones, debemos calificar la Infraestructura informatizada (IT).
La inclusión de un sistema informatizado en la manufactura no debe aumentar el riesgo total del proceso.
Estos lineamientos de la nueva GMP están basados en la GAMP5 (ISPE) A Risk-based Approach to Compliant GxP Computarized Systems.
Como parte del sistema de gestión de riesgos, las decisiones sobre la extensión de la validación y de los controles de la integridad de datos deben basarse en una evaluación de riesgos del sistema informatizado justificada y documentada.
La norma menciona diferentes figuras que deben existir, como: el propietario del proceso (process owner), el propietario del sistema (system owner), las Personas Calificadas e IT. Obviamente todo el personal debe estar calificado, con su nivel de acceso y definidas sus responsabilidades.
Los proveedores de sistemas informatizados deben estar evaluados y debe disponerse de acuerdos técnicos y acuerdos de confidencialidad (ppalmente. para acceso remoto). La necesidad de auditarlos debe estar basada en un análisis de riesgo del mismo.
Debe disponerse de un inventario de los sistemas computarizados del laboratorio.
La Validación debe ser proporcional al riesgo del sistema (clasificación del riesgo, por ejemplo Categoría GAMP5 y calificación del proveedor), a más riesgo más esfuerzos de validación.
La transferencia de datos debe ser efectuada de forma segura.
En cuanto a la Operación de los SC, debemos asegurar que los datos son guardados regularmente de forma integra durante el período de conservación de los mismos. Debe haber un Audit Trail y debe ser considerada la Gestión de cambios y configuración.
Los sistemas informatizados deben ser revisados periódicamente para verificar la validez de la validación. Esta revisión incluye por ejemplo documentos de operación, validación, cambios, eventos, accesos y manejo de back up, por ej.
También hace referencia a otros temas de importancia como el manejo de firmas electrónicas, plan de Disaster Recovery, etc.
Desde cGMPdoc les ofrecemos distintas alternativas como:
- Entrenamientos “In Company” para capacitar a su personal
- La implementación del proceso de CSV (Validación de Sistemas Computarizados), lo cual incluye la discusión del procesos internamente con el laboratorio, la confección de los SOPs necesarios, el entrenamiento del personal, la confección del PMV de sistemas computarizados y la ejecución de una validación a modo de ejemplo, de manera que Uds. puedan luego continuar con las actividades del Plan.
- La validación de un sistema computarizado
- Asesoramiento en la revisión de su proceso de CSV si lo necesita
- Pack de SOPs y documentos modelos sobre CSV
Espero que les resulte de utilidad.
La validación es mandatoria para los sistemas GxP.
El nivel de la validación dependerá de la configuración del software y de cómo el proveedor diseñó, desarrolló y testeó el sistema computarizado (hardware y software).
La evaluación del proveedor y la categoría del software (de acuerdo a la GAMP5) son claves en el proceso de análisis de riesgo e influenciarán en la determinación de cuanto esfuerzo será requerido para la validación del sistema computarizado.
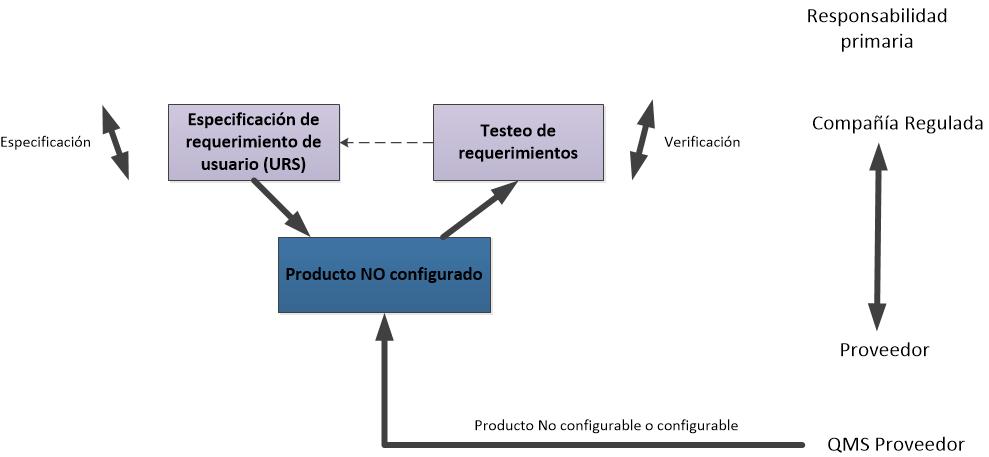
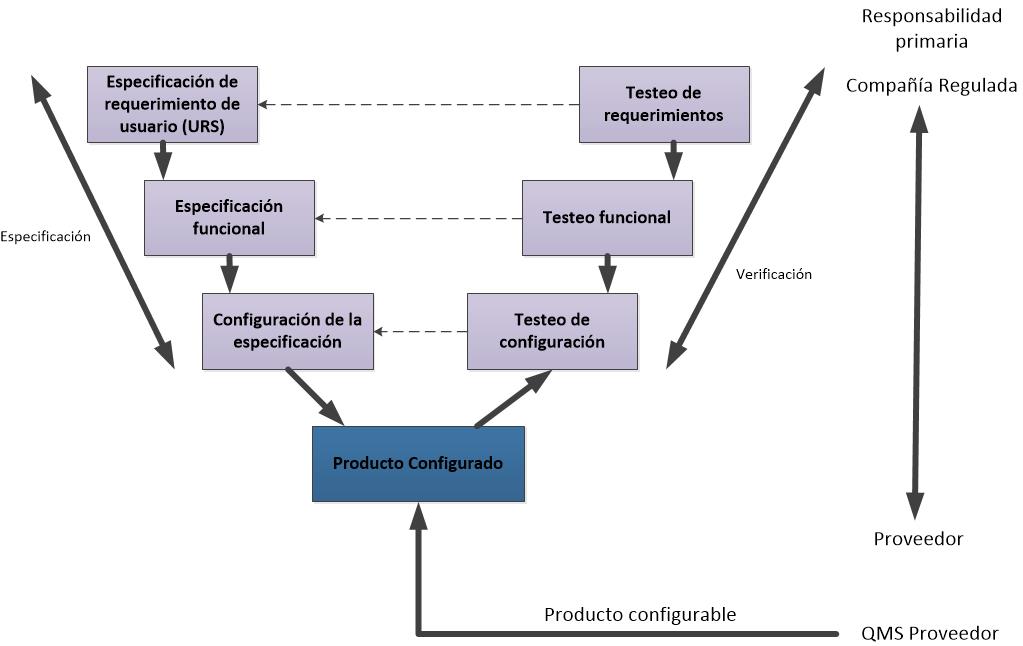
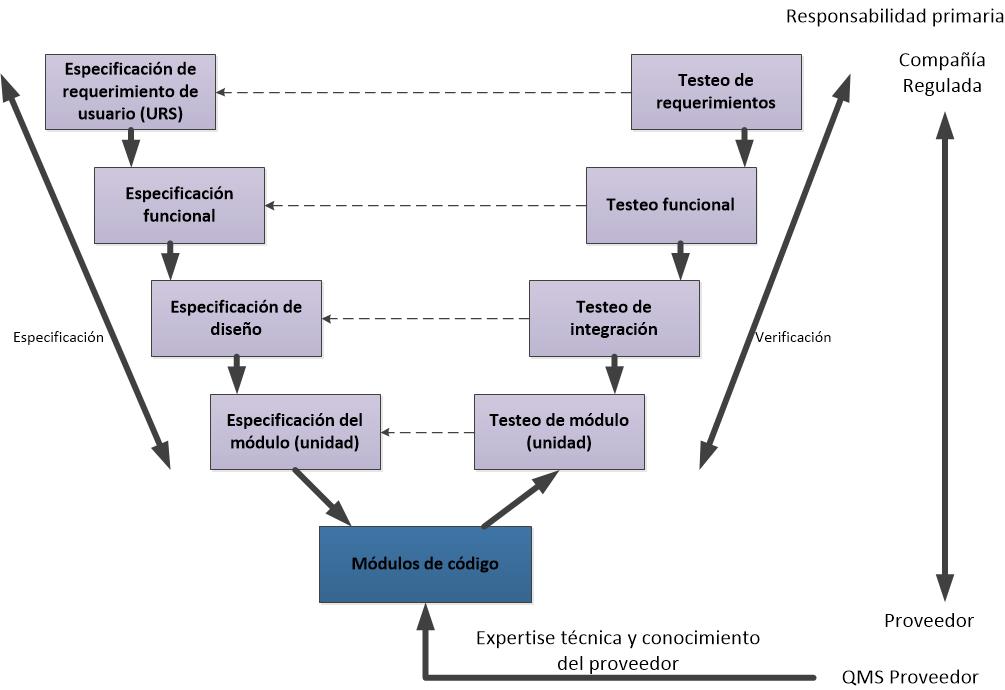
Les dejo los flujogramas propuestos para las actividades de validación de las categorías 3, 4 y 5, de acuerdo a la GAMP5 (2008).
Flujograma para un sistema de categoría 3.
Flujograma para un sistema de categoría 4 (adaptado).
Flujograma para un sistema de Categoría 5 (a medida).
¿Qué tipos de sistemas computarizados debemos considerar?
En principio los relacionados a las actividades de controles del proceso, los relacionados al laboratorio, las aplicaciones de IT y lo relacionado a infraestructura.
Espero que les haya resultado interesante.