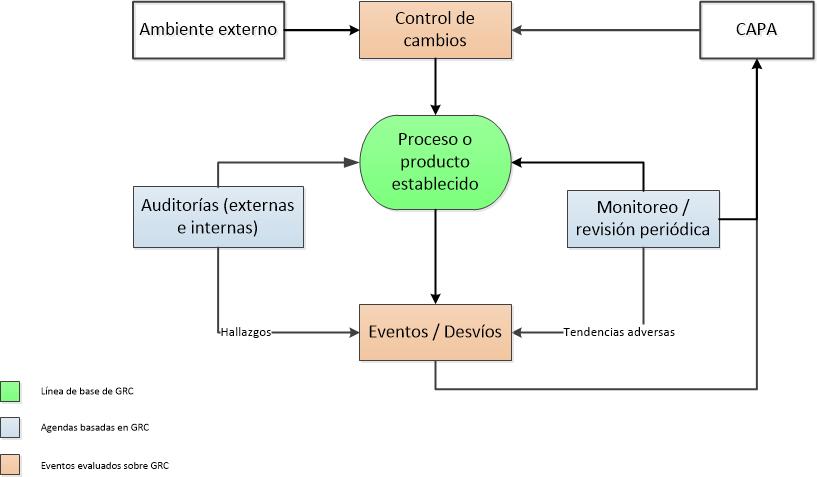
Durante bastante tiempo hemos tenido en mente que los procesos de validación y de calificación debían contemplar dentro de los procedimientos cuál era la frecuencia con que debíamos repetir la actividad, sin embargo las cGMP nos van orientando hacia implementar una serie de actividades sistemáticas que nos permitan revisar y documentar que nuestros equipos, procesos, sistemas, etc. mantienen el estado validado o calificado. Por ejemplo en sistemas computarizados hablamos de efectuar una revisión periódica de los sistemas computarizados validados, en el caso de los procesos validados hablamos de seguirlos a través de la verificación continua del proceso o de la Revisión Anual de Productos.
¿Cuáles son los ciclos adecuados para la recalificación de equipos?
Con la revisión del Anexo 15 en octubre de 2015, el tema de la recalificación se ha vuelto más importante. En el antiguo Anexo 15 de 2001, el tema de la recalificación estaba “oculto” bajo la demanda general de revalidación (punto 45). Con la revisión del Anexo 15, la recalificación ahora tiene su propio capítulo con los requisitos para:
- Una evaluación del equipo con la frecuencia adecuada para demostrar que se ha mantenido en un estado de control y
- Cuando es requerida una recalificación, los intervalos de tiempo deben justificarse y los criterios para la evaluación deben establecerse.
En la práctica, a veces es difícil establecer tales programas y criterios para la evaluación. En el área estéril, a veces hay referencias específicas de las regulaciones a dispositivos y procesos individuales. Por ejemplo, de acuerdo con la Guía aséptica de la FDA, los filtros HEPA en sala limpia clase 5 deben probarse dos veces al año. El Anexo 1 de las Directrices GMP de la UE también proporciona directrices sobre este tema (por ejemplo, para la esterilización).
¿Qué pasa con el equipo en otras áreas?
En este caso, puede ser útil el capítulo 9 de la línea de base ISPE n° 5 revisada Puesta en servicio y calificación de junio de 2019 sobre la revisión periódica. El enfoque de “revisión”, es decir, la evaluación, se presenta en dos fases.
En la fase 1, se clasifican los “sistemas de impacto directo” existentes. Dependiendo de la complejidad del sistema (complejo vs. estándar) y la influencia en la calidad del producto, los sistemas se dividen de manera ejemplar en las categorías 0-3. Cada categoría, excepto la categoría 0, se asigna a un período de “revisión”. La categoría 0 se refiere a los sistemas existentes donde tenemos disponibles datos de monitoreo. En este caso, no es necesaria una revisión de estos sistemas de categoría 0 (por ejemplo, sistemas de agua). Ya que tenemos datos para su evaluación. Para sistemas en la categoría 1, por ej. autoclaves, se aplican las especificaciones anteriores de las reglamentaciones estériles. Para sistemas de categoría 2, por ej. tanques de almacenamiento intermedio, la línea de base sugiere un intervalo de dos años. Para sistemas de categoría 3, por ej. comprimidoras, la línea de base recomienda un intervalo de revisión de tres años.
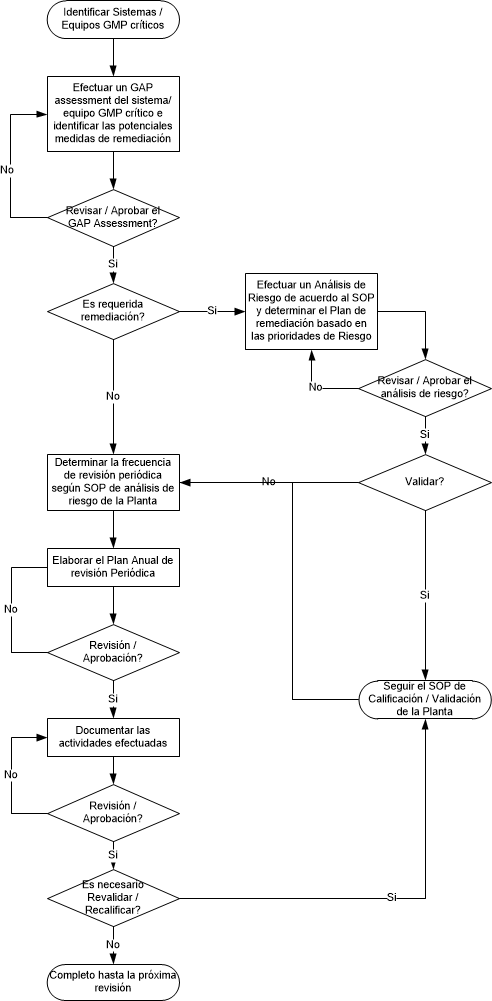
En la fase 2, se ejecuta la “revisión”. La revisión en sí tiene lugar como un proceso de tres pasos:
Paso 1: una evaluación inicial con respecto al cumplimiento de GMP, historial de cambios, mantenimiento / calibración y desviaciones.
Si esta evaluación plantea dudas sobre el impacto del sistema en la calidad del producto, se debe seguir el paso 2. De lo contrario, el proceso termina aquí.
Paso 2: en función del resultado del paso 1, los expertos en la materia (SME) examinan en mayor profundidad los sistemas examinados en el paso 1, cuyos resultados son cuestionables. Los SMEs deben provenir de los mismos departamentos que también participaron en la evaluación en el paso 1.
Si la evaluación en el paso 2 llega a la conclusión de que un sistema ya no puede estar en estado calificado, el siguiente paso, el paso 3, tiene lugar. De lo contrario, el proceso termina aquí.
Paso 3: en función del resultado del paso 2, los SMEs examinan en mayor profundidad los sistemas examinados en el paso 2, cuyos resultados son cuestionables. Los SMEs deben provenir de los mismos departamentos que también participaron en la evaluación en los pasos 1 y 2. Si es necesario, se deben tomar medidas para que el sistema vuelva a un estado calificado. Se debe crear un documento de desviación para controlar las acciones tomadas.
De acuerdo con la línea de base, todo el proceso debe ser administrado por alguien con una gran experiencia. La revisión en sí misma debe al menos ser aprobada por el propietario del sistema y la unidad de calidad. Un formulario de revisión es parte de la línea de base como Apéndice 11 y se da un ejemplo en el Apéndice 12.
Tomado de la news Letter de la ECA (28/8/2019)