El monitor de temperatura de un envío indicó, al recibirlo, que el medicamento excedió los 8 °C durante la duración del envío. El examen posterior encontró que el envío se acondicionó en la configuración correcta del contenedor de acuerdo con el procedimiento de operación estándar aprobado (SOP).
Todos los envíos anteriores se recibieron con el monitor de temperatura que indica que el producto se había mantenido dentro del rango de temperatura deseado de 2-8 ° C.
Una investigación reveló que la calificación del contenedor aislado y la configuración del pack de gel se había realizado de manera retrospectiva utilizando datos de tránsito reales para los envíos que habían ocurrido durante el invierno. Los datos de temperatura para los envíos de productos estaban dentro del rango y, por lo tanto, la configuración fue calificada. Cuando el envío se realizó a fines de la primavera, a una temperatura ambiente mucho más alta, la configuración no tenía suficiente capacidad de enfriamiento para mantener la temperatura y carecía de suficiente capacidad aislante para evitar la conducción de calor externo a la carga del medicamento. Por lo tanto, la validación carecía de una calificación formal que tuviera en cuenta todas las condiciones esperadas durante el tránsito para demostrar que la configuración del empaque cumplía con las especificaciones predeterminadas.
Los protocolos de validación para las cadenas de suministro de la cadena de frío deben requerir situaciones de exposición a temperatura máxima representativas del peor de los casos (Worst Case). Al desafiar el transporte de productos que requieren temperaturas moderadas controladas, se deben utilizar temperaturas altas en el peor de los casos (condiciones de verano) y temperaturas bajas en el peor de los casos (condiciones de invierno).
Posts tagged ‘Calificación’
El ISPE nos da algunos principios para efectuar las actividades de calificación:
- Enfoque en la calidad del producto: la calidad del producto debe ser garantizada por las actividades de calificación.
- Requisitos: el URS debe basarse en el proceso y esto se confirma en el PQ (calificación de performance). Esto significa que tanto el IQ como el OQ están subordinados en importancia.
- Evaluaciones de riesgo: el desarrollo del proceso y el diseño experimental son los elementos clave para identificar funciones y parámetros críticos, los cuales serán la base para las actividades de calificación.
- Actividades basadas en el riesgo, las mismas dependerán de la complejidad de la instalación (ej. Clasificación GAMP) y deben agregar valor a la calificación. No deberíamos incorporar solo actividades por el hecho de hacer volumen de documentación.
- Uso de la documentación del proveedor: Si es posible.
- Pruebas: por regla general, las pruebas deben realizarse una sola vez.
- Fomento de la innovación: en este punto, se requiere la flexibilidad de los programas de calificación para poder implementar nuevas tendencias.
Todo esto requiere grandes cambios organizativos en las empresas (quizás es una de las partes más difíciles), que a menudo tienen conceptos relativamente rígidos de calificación y validación.
La gestión de calidad se centraría más en los procesos y análisis de riesgo, los planes de calificación y los informes de calificación. Por otro lado, también sería necesario fortalecer los sistemas de calidad de los departamentos especiales (por ejemplo, Ingeniería, IT, etc.) y la producción. El riesgo para la calidad del producto se determinaría en el contexto de una divulgación de riesgo sobre la base de la comprensión del proceso. El control de esos riesgos se calificaría entonces en la calificación de la performance.
Muchas veces escuchamos esta pregunta, ¿Porqué debemos calificar un equipo, un sistema computarizado, una instalación o nuestro personal? Voy a tratar de mostrar el racional.
Por ejemplo frente a un problema, un desvío de manufactura o un resultado OOS, la investigación conduce a la identificación de la causa raíz. Por ejemplo si efectuamos un Ishikawa, identificamos un número de elementos de la manufactura o del sistema del laboratorio, donde la variación en al menos uno de esos elementos podría llevar al desvío.
Estos elementos pueden incluir al equipo, las instalaciones, los servicios, materias primas, procedimientos, performance del personal, etc. Cada uno de esos elementos es revisado, y se van eliminando, hasta que uno (o más) permanece y ese elemento es considerado como la causa raíz.
Un elemento es removido de esta lista una vez que es determinado que no podría haber sido la causa raíz del desvío. Es aquí donde el proceso de calificación se convierte en importante. Una excelente forma de eliminar un elemento de la consideración adicional como una causa raíz de un problema es por medio de la calificación del elemento por adelantado.
Tomemos un equipo, por ejemplo, el IQ asegura que el mismo ha sido instalado dentro de las especificaciones del diseño. El OQ asegura que el equipo opera de acuerdo a las especificaciones de diseño y el URS (especificaciones de requerimiento del Usuario) y finalmente el PQ asegura que el equipo muestra una continua adecuación para sus fines de uso.
El IQ, OQ y PQ son críticos para los sistemas farmacéuticos, biotecnológicos y dispositivos médicos del laboratorio. Las Compañías deben instalar, operar y mantener los equipos utilizados en la elaboración y control de sus productos dentro de especificaciones, asegurando que sus operaciones son confiables y la calidad del resultado o producto es consistente.
Dado que no ha habido variaciones en el equipo desde que se ha calificado, no puede ser la causa raíz de la desviación de manufactura o del resultado OOS del laboratorio. A través del proceso de calificación, este elemento puede ser eliminado de la consideración en la investigación.
Este mismo enfoque puede ser aplicado para la performance del empleado. Al mismo tiempo los empleados que trabajan sobre el proceso que ha generado la desviación han sido entrenados sobre los SOPs pertinentes (o no), tipo de entrenamiento, entrenadores, material del entrenamiento, verificación de la efectividad del mismo, etc.

Cada elemento constituyente es considerado y eliminado de la consideración cuando es determinado que este no ha sido la causa raíz de la desviación. El proceso de calificación del empleado provee una importante forma de eliminar un elemento constituyente por adelantado. Estaba el entrenador calificado? Fueron los empleados calificados?
Esta discusión ha considerado el racional para calificación, resaltando el role que el proceso de calificación juega en las investigaciones de desvíos y análisis de causa raíz. La calificación del empleado prueba ser un tipo de entrenamiento relativamente caro cuando se lo compara con el entrenamiento convencional.
Cómo determina una organización cuáles procedimientos / procesos requieren calificación del empleado, y cuales solo requieren entrenamiento convencional? Esto plantea la cuestión de la criticidad del procedimiento.
Procedimientos complejos y críticos requieren calificación del empleado, requieren un alto nivel de habilidad, alto nivel de seguridad del empleado, o puede resultar en un riesgo de cumplimiento del negocio de la compañía si no es efectuado apropiadamente.
Esto usualmente requiere de un análisis de riesgo.
Si un procedimiento es o no considerado crítico debería guiarse por medio de tres preguntas:
- ¿Qué podría salir mal con el proceso asociado?
- ¿Cuál es la probabilidad que esto suceda?
- ¿Cuál es la severidad de las consecuencias si esto sucede mal?
De acuerdo a las ICH Q9 Gestión de riesgos de calidad, el nivel de esfuerzo, la formalidad y documentación del proceso de GRC debe ser conmensurado con el nivel de riesgo.
En el primer caso, cuando el proceso o procedimiento es crítico es esperado que el personal sea calificado en el mismo, y en los procesos de menor complejidad puede ser requerido un entrenamiento convencional, con evaluación del aprendizaje. Las situaciones intermedias deben ser evaluadas por el management, quién finalmente decidirá el tipo de entrenamiento requerido.
Por supuesto, el criterio y el proceso de decisión para seleccionar el tipo de entrenamiento debe ser incorporado a un procedimiento escrito.
La construcción de estos bloques nos permitirán atar el cumplimiento y la calidad al proceso de validación:

- Conocer el proceso (es clave)
- Evaluar los riesgos de fabricación al producto
- Identificar los controles y las estrategias de control incorporados en el diseño
- Implementar los controles de riesgo de los aspectos críticos
- Efectuar la calificación / validación (documentación)
- Manejar el concepto de ciclo de vida, NO de evento
- Efectuar la revisión continua y la mejora en base a datos de calidad – escuchar la “voz del proceso”
Espero que les resulte útil.
El proceso de auditoria de integridad de datos al laboratorio, requiere de manejar algunos principios, además de un manejo del proceso.
A modo de ejemplo les dejo este esquema de un proceso de HPLC, para que puedan considerar los elementos a evaluar desde el punto de vista de integridad de datos, y además a continuación del mismo, algunos temas a considerar para efectuar la auditoría de integridad de datos, espero que les resulte útil.
NOTA: no considere la preparación de muestra y standars.
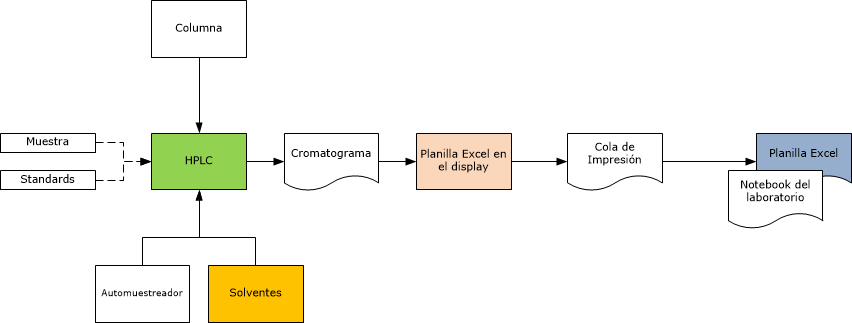
Auditoría de Integridad de datos
- Podríamos decir que cada paso en cualquier proceso de laboratorio tiene un potencial problema de integridad de datos, después de todo los datos son un producto del laboratorio, es como una fábrica de datos
- La herramienta clave del auditor cuando audita la integridad de datos, es el Audit Trail.
- Si hay un Audit trail electrónico disponible, debe ser seguro de auditoría electrónica está disponible debe ser seguro (no susceptible de ser cambiado), accesible, imprimible e inequívocamente asociada con una muestra de análisis en particular.
- Si no hay un Audit trail electrónico disponible, entonces como alternativa se debe trazar todas las actividades a través de las etapas, buscando vínculos entre las etapas y verificando la existencia de todos los documentos o archivos de soporte.
- Otra herramienta es la secuencia de números de análisis, automáticamente asignada a cada inyección (por ej.) en un HPLC. No debe haber archivos perdidos, aunque algunos de ellos serán sólo para propósitos de set-up / preparación.
- En la práctica, hay que seleccionar la más importante para el rastreo de datos para poder efectuar una investigación detallada y siempre incluir cualquier transcripción manual de datos, por ej: ¿Fue correcta? ¿Fue revisada? ¿Cuándo y por quién? ¿Dónde está la evidencia?, etc.
- Desde el punto de vista electrónico, ¿Las planillas de cálculo están controladas? ¿Cómo? ¿Cómo son manejados los cambios de las planillas de cálculo? ¿Fue usada la versión correcta? ¿Está validada? ¿Dónde está el informe? ¿Dónde está la impresión de los resultados (si se hace)?, etc.
Para todos aquellos interesados, envíennos sus respuestas, comentarios o dudas, a info@cgmpdoc.com y con gusto les compartiremos nuestra solución al tema.
Hay dos componentes necesarios para que una auditoría sea exitosa.
El primero es un auditor con las habilidades, la educación y la experiencia correcta o necesaria (ver haciendo click aquí).
El segundo es el proceso de auditoría por sí mismo.

El proceso de auditoría puede comenzar algunos meses antes de que la auditoría se lleve a cabo. Las etapas del proceso de auditoría son:
Preparación de la auditoría
- Determinar quién conducirá la auditoría – un individuo o un equipo.
En el caso de un equipo, el auditor líder será responsable de asegurar que la auditoría es manejada efectivamente y eficientemente.
- Desarrollar la agenda de la auditoría
Para conducir una auditoría eficiente y efectiva, debe ser elaborada una agenda. Esto ayudará a mantener el flujo de la misma sin problemas y hará mejor el tiempo del auditado y del auditor.
El propósito de la auditoría debería estar claro para ambos, el auditado y el auditor. Puesto que efectuará una auditoría GMP, Ud. Debería considerar los siguientes temas GMP:
- Sistemas de Calidad
- Gestión de materiales
- Controles de calidad del laboratorio
- Sistemas de producción
- Instalaciones y equipos
- Empaque y rotulado
Durante la auditoría el auditor debe planear prestar especial atención a los sistemas de calidad, la forma en la cual el laboratorio maneja los desvíos, los reclamos y los resultados de los ensayos OOS (fuera de especificaciones), esto le dará una visión de la cultura de calidad de la compañía y su adherencia a las cGMPs.
- Confirmar la auditoría y acordar una fecha con el proveedor /área a ser auditada.
Trabajar con el auditado para determinar un tiempo para ajustarse dentro del marco de tiempo que sea conveniente para ambas partes.
- Revisar documentos, incluir documentos recibidos desde la empresa o el laboratorio
- Preparar notas para la auditoría
Luego de revisar la información de soporte y los documentos de referencia y los estándares, el auditor prepara notas que lo ayudará a efectuar la auditoría, así como una lista de documentos a revisar.
Potenciales actividades de la auditoría en el laboratorio
Todas las auditorías deben ser conducidas de una forma abierta, el auditor debe discutir los hallazgos y expectativas con el auditado mientras conduce la auditoría. La auditoría es una oportunidad de identificar oportunidades de mejora para el auditado.
- Conducir una reunión de apertura
Al arribar al laboratorio, el auditor / equipo auditor debe efectuar una reunión de apertura con los responsables del laboratorio (QA, producción, dirección, otros representantes asignados). Durante la reunión de apertura, el auditor debe asegurarse que el auditado entienda su rol en la auditoría.
- Recorrer los depósitos, instalaciones de manufactura y laboratorios, si es apropiado.
Las auditorías pueden comenzar con una recorrida a las instalaciones y al laboratorio, es recomendable “seguir un producto” (desde la recepción de materiales hasta la liberación del producto terminado).
Durante la recorrida, verificar que los SOPs e instrucciones están siendo seguidos y los registros se completan con exactitud.
A medida que realiza la auditoría el auditor puede seguir las notas preparatorias, para asegurar que cubre la auditoría completa.
Las observaciones de la auditoría pueden ser clasificadas en críticas, mayores y menores.
Si un tema o desvío crítico es observado, el mismo debe ser comunicado inmediatamente al management del laboratorio y al responsable auditado
- Entrevistar analistas y operadores (si es necesario).
- Efectuar una reunión informativa de la auditoría con el auditado, si es apropiado.
Para las revisiones informativas donde las observaciones y hallazgos deberían ser revisadas con el auditado como también las áreas de preocupación para asegurar que no habrá sorpresas o malos entendidos en el cierre final de la auditoría.
La agenda de la auditoría puede ser actualizada si son requeridos cambios basados en las observaciones o recursos.
- Efectuar reuniones de auditoría con el equipo, si un enfoque de equipo auditor es utilizado.
Estas reuniones son mantenidas para ayudar al equipo auditor a mantenerse enfocado en el objetivo de la auditoría y la auditoría en curso.
- Revisar la documentación del auditado
Esto puede incluir SOPs, documentación de manufactura y de ensayos, datos crudos asociados, protocolos de calificación y validación, y reportes de investigaciones.
- Conducir la reunión de cierre
La reunión de cierre es el foro usado para presentar verbalmente los resultados de la auditoría al management auditado. Es esencial que todos los participantes claves de la auditoría y el management del auditado participen de la reunión de cierre.
Seguimiento de las actividades de la auditoría
- Documentar las observaciones en un reporte formal
Los mismos están basados en las anotaciones que el auditor tomó durante la auditoría. Los mismos pueden ser usados por diferentes personas para diferentes motivos, por eso necesitan ser claros y escritos de una forma consistente como también contener la información relevante y necesaria.
Los reportes de auditoría deben ser normalmente emitidos dentro de un cierto plazo, así como también las respuestas conteniendo el plan de acción.
- Evaluar las respuestas recibidas desde la empresa / laboratorio
Luego que las respuestas son recibidas, es necesario analizarlas y determinar si las acciones correctivas planteadas y los riesgos propuestos son aceptables. Una vez recibida la respuesta, y si las respuestas son consideradas aceptables, el auditor tiene la responsabilidad de responder dentro de un período de tiempo definido.
- Cerrar la auditoría
- Efectuar el seguimiento de los CAPAs generados
El seguimiento puede requerir nuevas visitas al laboratorio o la recepción de las evidencias de las acciones cumplidas, para darle cierre a la acción pendiente.
Evaluación de riesgo durante el proceso de auditoría:
El auditor toma decisiones basadas en el riesgo a través del proceso de auditorías, por ejemplo dado que No es posible cubrir todo durante el proceso de auditoría, es necesario evaluar los riesgos, tal vez efectuar un análisis de riesgo formal y hacer un juicio sobre que cubrir. Este juicio que necesita hacer puede necesitar cambiar durante el proceso de auditoría basado en nuevos conocimientos.
Herramientas de análisis de riesgos:
Para soportar la evaluación de riesgo de un proveedor, el auditor debe elegir usar una herramienta de análisis de riesgo. Una de las herramientas formales de análisis de riesgo usadas por los auditores es Failure Modes Effects Analysis (FMEA).
Si Ud. está interesado en entrenar a su equipo auditor, consulte en info@cgmpdoc.com. Podemos ofrecerle materiales de entrenamiento para Auditores, check lists para que Ud. Efectúe las distintas auditorías y además tenemos la posibilidad de entrenar a su equipo “In Company”.