Dentro de estos requerimientos básicos, les dejamos 10 tips claves, esenciales para asegurar la integridad de los datos dentro de los límites de un sistema de datos electrónicos:

- Identidad única para cada usuario:
Para sistemas basados en papel, siguiendo las Buenas Prácticas de Documentación la identificación de cada analista puede ser alcanzada a través de que cada persona firme o coloque sus iniciales sobre una impresión o notebook de laboratorio. Con los sistemas electrónicos, este principio debe ser mantenido por medio de la identidad única del usuario.
Las cuentas de usuarios no deben ser compartidas, por ej. Una cuenta de usuario establecida para dos analistas para el acceso a un software de laboratorio de un sistema HPLC.
Las identidades de los usuarios no deben ser reusadas, si una persona deja el laboratorio o la compañía, esa identidad del usuario (ID) debe ser removida del uso y cualquier nuevo usuario debe ser habilitado con un nuevo y único número de cuenta de usuario.
- Implementar controles de passwords adecuados:
Los passwords deben ser efectivos de manera que no puedan ser adivinados por otros, por ej. Su nombre / dirección / fecha de cumpleaños, etc. pero no lo demasiado complejos de manera que el usuario lo tiene que tener escrito para poder recordarlo.
Los passwords NO deben ser compartidos bajo ninguna circunstancia. Cuando las credenciales de registro son compartidas, los individuos específicos no pueden ser identificados a través de la actividad de registro y por consiguiente se pierde la integridad de datos.
- Establecer diferentes tipos de usuarios con diferentes privilegios de acceso:
Los accesos a los sistemas o equipos deben ser limitados solo a individuos autorizados, por ej. Analistas, administradores.
Cada tipo de usuario debe ser asignado con diferentes privilegios de acceso de acuerdo al rol que efectuará en el sistema. Debe haber protocolos de seguridad de datos en los cuales se describe el rol del usuario y sus responsabilidades en términos de privilegios de acceso, cambio, modificación, creación y eliminación de proyectos y datos. Solo personal senior debe tener cuentas que le permiten administración completa del sistema, incluyendo la edición de los métodos y de los proyectos.
El rol del administrador del sistema debe asegurar que cualquier requerimiento de cambio, eliminación, etc. es cuidadosamente registrado, y atribuido a un individuo para asegurar que la trazabilidad es mantenida.
- Establecer y mantener una lista de los usuarios actuales e históricos:
Una lista de los usuarios del sistema actuales y de los previos necesita ser establecida y mantenida, si un miembro del personal ha dejado de serlo, entonces la cuenta debe ser inmediatamente puesta como no disponible.
- Control de cambios del sistema:
Relacionado con la asignación de privilegios de accesos está la capacidad de un usuario de hacer cambios. Los cambios deben ser solo efectuados por medio de los individuos autorizados, ej. Cambios de métodos, parámetros de integración y línea de base. Esto debe ser posible para identificar cuáles individuos hicieron los cambios para determinar si ellos tienen el apropiado entrenamiento, educación y experiencia.
- Solamente personal entrenado debe operar el sistema:
Hay un requerimiento para que todo el personal tenga la combinación de educación, entrenamiento y experiencia para efectuar su trabajo. Una parte clave de cualquier programa de entrenamiento es que debe hacer foco sobre la generación de datos y que los cambios solo pueden ser hechos de acuerdo a procedimientos predefinidos para prevenir una acusación de falsificación o fraude.
Los usuarios de instrumentos o equipos deben poder demostrar su capacidad de uso frente a las agencias regulatorias cuando es requerido, dentro de su descripción de rol.
- Registrar toda la información
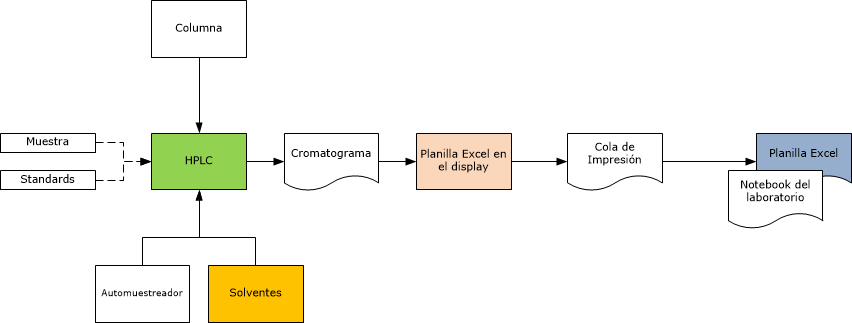
Los registros de laboratorio deben incluir datos completos derivados de todos los ensayos necesario para asegurar el cumplimiento con las especificaciones y estándares establecidos. Esta es la clave para establecer la integridad de los datos en un sistema de captura de datos electrónicos, a pesar de las situaciones que nos pueden ocurrir, errores, mal funcionamiento de un equipo, etc.
Los datos crudos, “raw data”, (ej. Cromatogramas, pesadas del estándar y de muestras, cálculos, estándares, reactivos, información del instrumento) deben estar disponibles siguiendo la ejecución del trabajo para permitir que una investigación pueda ser llevada a cabo eficientemente y para confirmar la autenticidad y confiabilidad de los datos durante la inspección efectuada por la agencia regulatoria. Todos los datos deben ser registrados de manera que las actividades puedan ser cuidadosamente reconstruidas.
- Definir y documentar registros electrónicos para el sistema
Para registros electrónicos los usuarios deben determinar cuáles datos serán definidos como “raw data”, como mínimo, todos los datos sobre los cuales se basa la toma de decisiones de calidad deben ser definidos como raw data.
Toda la documentación creada durante el análisis la cual incluye un registro es considerada como evidencia de una actividad, de este modo, los archivos de datos creados durante una corrida analítica son registros.
Muchos documentos (instrucciones y/o registros) pueden existir en formatos híbridos, por ej. Algunos elementos como electrónicos y otros en papel. No importa si un registro es generado en papel, existe con firmas manuscritas y le sigue una impresión de un registro electrónico (sistema híbrido) o si es mantenido completamente electrónico.
Controles apropiados deben están en uso para asegurar la integridad de los registros a lo largo del período de retención. Debe estar claramente definido cual registro está relacionado con cada actividad de ensayo y donde es localizado el registro. Controles de seguridad deben estar en uso para asegurar la integridad de los registros a lo largo del período de retención y validado cuando sea apropiado.
- Revisión de las entradas al audit trail para cada lote
Parte de los datos completos de un sistema de datos cromatográfico en las corridas analíticas incluyen el audit trail, y hay un requerimiento bajo las cGMP para la firma o iniciales de una segunda persona para mostrar que el trabajo ha sido hecho correctamente y conforme a los estándares.
Los cambios no deben tapar los datos registrados previamente (sobre escritura).
- Copia de seguridad del Sistema regular
Copias de seguridad (BackUp) regulares de toda la información relevante debe ser llevada a cabo. Típicamente esto se hace diariamente. La integridad y la exactitud de los datos de back up y la capacidad para recuperarlos datos debe ser verificada durante la validación y monitoreada periódicamente. Cuando el back up es efectuado, los registros del back up deben ser chequeados para ver cuando fue efectivo. El back up necesita ser validado, y periódicamente debe efectuarse una recuperación de los datos para ver que el hardware, por ej. CDs, son aún leíbles.