“Guía estándar para el desarrollo y la validación de procesos de limpieza basados en la ciencia y en los riesgos”
En 1996 la FDA tenía la expectativa que los elaboradores identificaran cualquier droga que produjera un riesgo de contaminación cruzada y que se implementaran las acciones necesarias para eliminar el riesgo. Los Subject Matter Experts (SME) desafiaban los límites de la reducción 1/1000 de la dosis terapéutica y/o < 10 ppm del lote de menor tamaño. En algunos casos esos límites no fueron lo suficientemente bajos y más importante aún no consideraban datos toxicológicos de los productos.
Hace ya algunos años, fue establecido un nuevo enfoque, basado en los HBEL (límites de exposición basados en la salud), para los trabajadores farmacéuticos expuestos a las drogas, en lugar de los criterios anteriores. Fue utilizado el ADE (Exposición diaria aceptable), definido como una dosis que es poco probable que cause un efecto en un adulto, si el individuo está expuesto por cualquier ruta a esa dosis o menos cada día por el resto de su vida.
La ASTM E3106 se apoya en los principios de la Guía de validación de procesos de la FDA y de la Gestión Riesgos de Calidad (GRC) de la ICH Q9, para implementar el enfoque de Riesgo.
Flujo del proceso
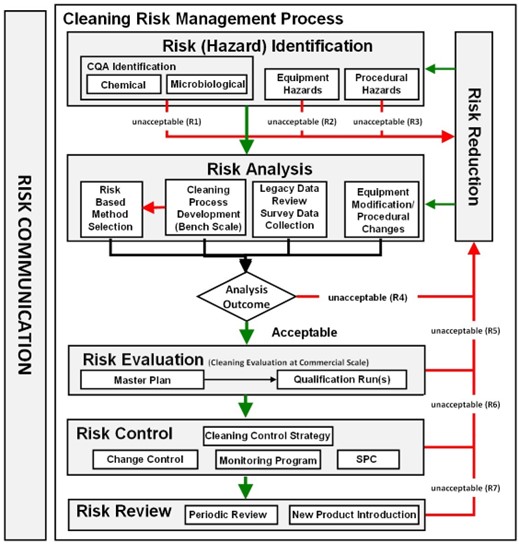
Los conceptos de la Guía de validación de procesos de la FDA son perfectamente aplicables a la validación de limpieza:
- Diseño del proceso de limpieza. Captura de conocimiento. Uso de herramientas de Risk Assessment para detectar variables potenciales. Por ejemplo mediante un FMEA, se debe determinar las posibles fallas y sus riesgos, y tomar acciones necesarias para mejorar los SOPs y lograr que sean robustos y confiables. Considerar los diseños de los equipos y además seleccionar los métodos analíticos adecuados, en base a la ciencia y el riesgo. La técnica debe ser simple y con bajo error potencial, a mayor riesgo del residuo, mayor sofisticación de la misma. La técnica debe poder detectar niveles menores a los permitidos en las muestras de limpieza. Los estudios pueden ser efectuados a pequeña escala inicialmente.
- Calificación de la limpieza. Usar herramientas estadísticas para analizar los datos colectados. Determinar el CpU de los residuos de limpieza. Si la capacidad del proceso es muy baja, debe ser estudiado y mejorado el proceso de limpieza, hasta asegurar que los residuos son efectivamente removidos y estan en niveles aceptables.
- Mejora continua. Usar los datos históricos (monitoreo) o avances tecnológicos para mejorar el proceso de limpieza. La estrategia de control puede incluir el monitoreo de parámetros críticos del proceso y muestreo y testeo de atributos de calidad y sistemas críticos o ser solo una inspección visual en situaciones de bajo riesgo.
Respecto de la ICH Q9, riesgo es:
R = S del peligro (Toxicidad del producto) x P de presencia de residuos o exposición al peligro x D o Detección del residuo
Alto riesgo (bajo HBEL, dificultad para limpiar y dificultad para detectar).
Esta situación requerirá mayores esfuerzos y alto nivel de documentación y debe estar alineada con los principios de la ICH Q9, donde el nivel de esfuerzo, formalidad y documentación debe ser proporcional al nivel de riesgo.
Si el riesgo no es aceptado, deben ser efectuados más esfuerzos para disminuirlos hasta su aceptación.
Cuando no pueden ser alcanzados resultados satisfactorios, el equipo / tren puede necesitar ser modificado, reemplazado o dedicado.
Una matriz basada en la toxicidad de los activos y la capacidad del proceso de limpieza proveerá un medio de selección de una estrategia de control basada en los principios de GRC (ICH Q9), por ejemplo, la Matriz de Shirokizawa:
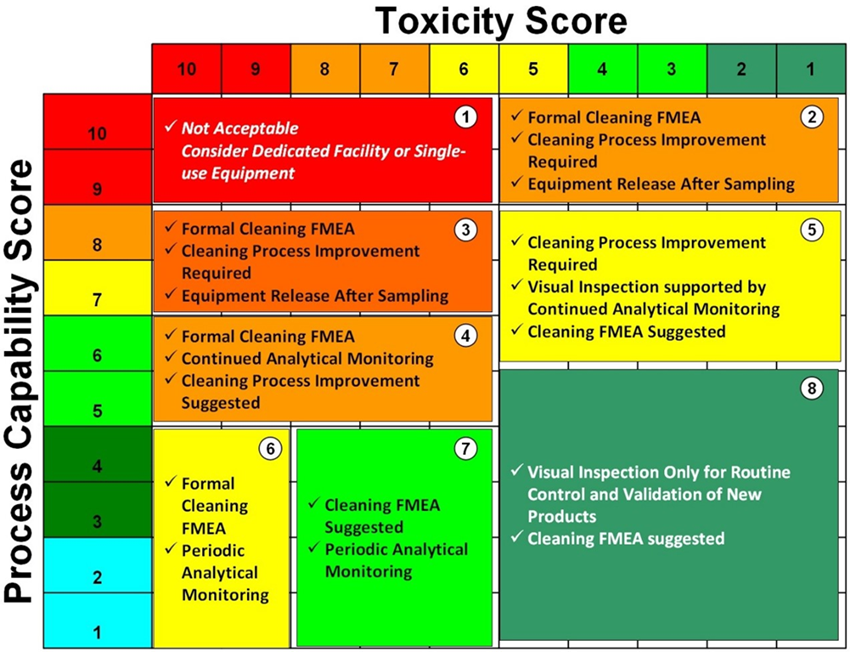
La Escala de capacidad = (1/CpU) x 10. La escala de Toxicidad esta dada por los HBLEs.
Cambios
Este proceso debe ser revisado básicamente cuando hay cambios en el proceso de limpieza o cuando es incluido un nuevo producto dentro del tren de elaboración, en este último caso debe ser determinado el nuevo HBEL del API, luego la capacidad de limpieza del proceso (a escala laboratorio), si el nuevo producto NO es removido adecuadamente con el SOP actual, debe ser efectuado el desarrollo de un nuevo método de limpieza y efectuados los estudios de validación, caso contrario no es necesario efectuar actividades adicionales.
