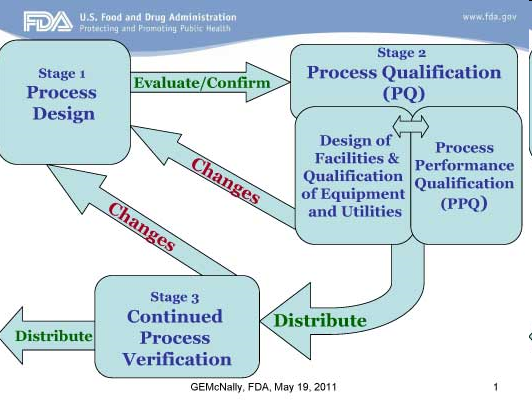
¿Qué influencia pueden tener los cambios de proceso en la validación del proceso?
¿De qué se trataba? Para ahorrar tiempo de producción, un fabricante de APIs había realizado un cambio de proceso en 3 puntos importantes de dos APIs. Después del cambio de proceso, también se produjeron lotes de Calificación del rendimiento del proceso (PPQ). Sin embargo, algunos de estos lotes no cumplieron con la especificación sobre uniformidad de mezcla. Los otros lotes fueron liberados.
La FDA comentó en la Warning Letter que el fabricante carecía de datos que mostraran que el proceso estaba en “estado de control” antes de la liberación del lote.
La evaluación del fabricante después del cambio de proceso mostró que el nuevo proceso conducía a deficiencias en la uniformidad de la mezcla. El proceso “antiguo” era más consistente y robusto. A este respecto, los lotes inéditos deben continuar retenidos durante la discusión sobre cómo proceder con el cambio de proceso. Después de 8 meses, se volvió al proceso “antiguo”. Sin embargo, el fabricante decidió que los lotes retenidos podrían distribuirse. Esto fue criticado por la FDA. El fabricante respondió que “revisaría” estos lotes y los retiraría si fuera necesario. En su respuesta también escribió que sólo los primeros lotes producidos consecutivamente “con éxito” son elegibles para el lanzamiento al mercado y que los lotes de PPQ no conformes se consideran de antemano. Si se observa un error en los lotes de PPQ y no hay una “causa raíz” clara para la causa, estos lotes no deberían recibir el lanzamiento de mercado.
A pesar de esta respuesta del fabricante, la FDA considera que la estrategia de validación del fabricante es insuficiente. Esto se debe a que la estrategia de validación no garantiza que los lotes de PPQ se evalúen correctamente para indicar que el proceso está en “estado de control”.
La FDA también señala que se le informó que el fabricante recientemente tomó la decisión de retirar todos los lotes de PPQ producidos con el nuevo proceso. Sin embargo, la FDA desea ver:
- una descripción detallada del programa de validación que garantiza que el estado de control se mantenga durante todo el ciclo de vida del producto
- instrucciones de trabajo adjuntas
- una descripción de la Calificación del rendimiento del proceso y un seguimiento adicional de la variabilidad intralote e interlote
- una descripción de si se ha realizado correctamente una PPQ para cada producto comercializado con los criterios adecuados
Puede encontrar la Warning Letter completa en el sitio de la FDA.
Tomado de Boletín ECA GMP 12/dic/2020


{kind=link}