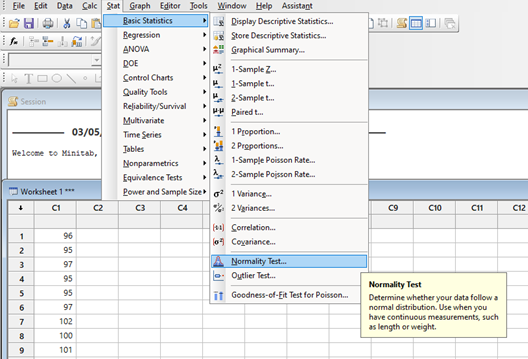
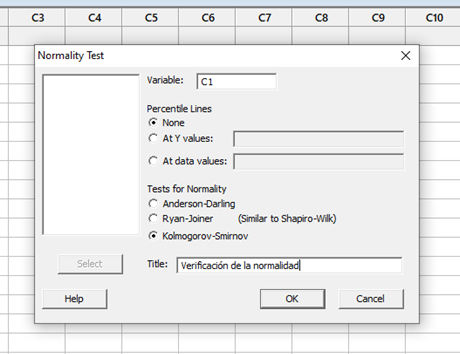
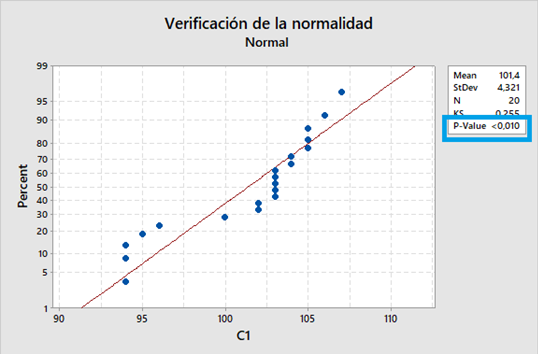
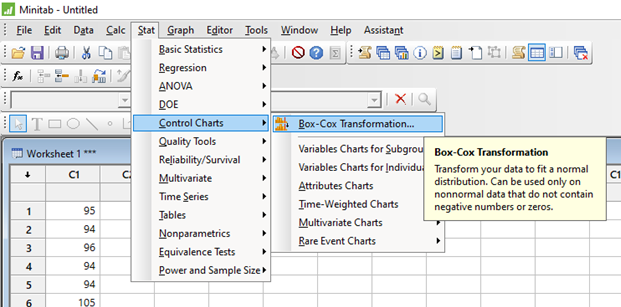
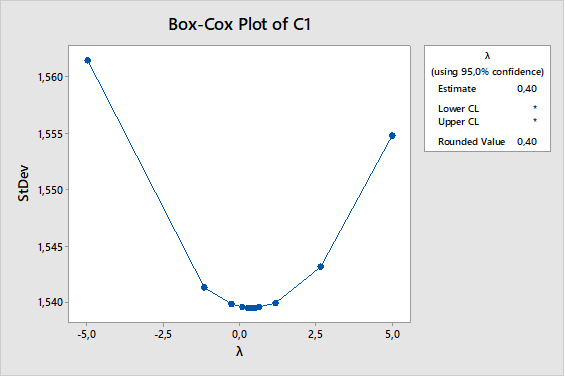
El diseño de experimentos (DOE) es un enfoque estadístico para la optimización de productos y procesos. Consiste en el estudio de distintas situaciones experimentales, que tienen sobre ciertas respuestas cuantitativas de unidades experimentales en observación.
De acuerdo a la definición de Wikipedia, el diseño experimental es una técnica estadística que permite identificar y cuantificar las causas de un efecto dentro de un estudio experimental. En un diseño experimental se manipulan deliberadamente una o más variables, vinculadas a las causas, para medir el efecto que tienen en otra variable de interés. El diseño experimental prescribe una serie de pautas relativas qué variables hay que manipular, de qué manera, cuántas veces hay que repetir el experimento y en qué orden para poder establecer con un grado de confianza predefinido la necesidad de una presunta relación de causa-efecto.
En la Industria farmacéutica actualmente es de suma importancia, constituyendo una parte sumamente importante del desarrollo de productos y la mejora continua del proceso.
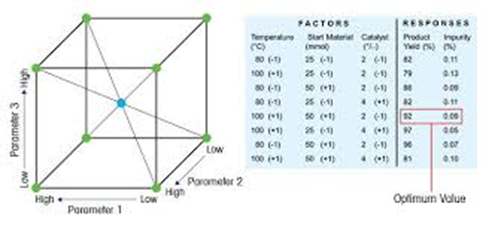
¿Qué ventaja aporta el DOE en el desarrollo farmacéutico?
Nos da la posibilidad de evaluar simultáneamente uno o varios parámetros de entrada, o “factores”, como la temperatura, la materia prima o la concentración, para determinar las condiciones en las que los atributos del producto, o “respuestas”, como el rendimiento, la selectividad o el nivel de impurezas, alcanzan un valor óptimo.
Eventos o pasos del DOE
Todo experimento involucra una serie de eventos:
1.Conjetura: hipótesis original que motiva el experimento ej. Marca de nafta vs rendimiento del coche.
2.Experimento: prueba efectuada para mostrar la conjetura.
3.Análisis: análisis estadístico de los datos obtenidos del experimento.
4.Conclusión: lo que se ha aprendido de la conjetura original con relación al experimento. A menudo conduce a una conjetura nueva.
En un próximo artículo daremos un ejemplo práctico del uso del DOE.