En Julio 2022, fue publicada la segunda edición de la Guía ISPE GAMP® 5: un enfoque basado en el riesgo para sistemas computarizados compatibles con GxP.
Esta segunda edición de ISPE GAMP 5 resalta la importancia de los proveedores de servicios, del mayor uso de herramientas de software y automatización para lograr un mayor control, una mayor calidad y menores riesgos a lo largo del ciclo de vida y además destaca la importancia del “pensamiento crítico”.
En este punto quiero detenerme y dejarles un resumen de un artículo escrito por Charlie C. Wakeham, sobre Por qué necesitamos el pensamiento crítico:
Puede parecer que las únicas cosas que la industria farmacéutica ama más que los acrónimos son las nuevas frases de moda. Y eso puede ser verdad. Pero a veces, detrás de la frase de moda, hay beneficios reales, fuertes y alcanzables para nuestro trabajo, nuestra industria y nuestros pacientes finales.
El pensamiento crítico es una de esas frases de moda. Originadas fuera de nuestra industria y adoptadas con entusiasmo por instituciones académicas y consultores a nivel mundial, la mayoría de las definiciones son algo ininteligibles y difíciles de relacionar con la vida real.
Charlie menciona que hay no menos de 11 definiciones, pero me voy a quedar con la que para él es su propia definición:
Es elegir sabiamente, excluyendo la burocracia, la jerarquía, el ego, el sesgo y la ambición del proceso de toma de decisiones.
Se está enfocando en lo que importa, que tiene que ser la calidad en lugar del cumplimiento y la documentación. Obtenga la calidad correcta y el cumplimiento estará allí de todos modos.
Me parece más importante lograr que la industria utilice el pensamiento crítico que debatir las complejidades del paradigma.
Veamos algunos ejemplos de pensamiento crítico con sistemas computarizados GxP:
ESPECIFICACIONES DE REQUISITOS
Todos estamos de acuerdo en que necesitamos capturar lo que debe hacer un sistema computarizado cuando estamos planeando un nuevo sistema, es decir, la funcionalidad que proporcionará para respaldar un proceso de negocios en particular. Esto significa que debemos definir qué regulaciones se aplican y qué requisitos individuales deben cumplirse dentro del marco regulatorio general. Necesitamos requisitos para definir la funcionalidad específica que el sistema debe proporcionar en relación con el proceso comercial; cómo controlará una actividad, analizará una muestra, calculará un resultado y almacenará los datos. Puede haber restricciones particulares o requisitos de infraestructura: alojamiento en la nube, funcionamiento en una sala limpia, conexión o interfaz con un sistema existente.
Todos estos son tremendamente importantes y deben ser capturados como requisitos.
Requisitos sobre cuántos niveles de submenú se permiten en el sistema. Requisitos para “rápido, fácil, fácil de usar”. Estos no tienen sentido para el proceso comercial e inútiles para el paciente final.
¿Qué hay de capturar los requisitos en un documento de Word en lugar de utilizar una herramienta de gestión de requisitos? ¿Qué elección de logotipo, fuente, interlineado? ¿Serán los requisitos firmados a mano o aprobados electrónicamente? Nada de esto tiene ningún impacto en la calidad del producto o la seguridad del paciente. Siempre que los requisitos estén controlados, aprobados, protegidos contra cambios no autorizados, disponibles y actualizados durante toda la vida del sistema, el formato, los medios y la apariencia de la información tampoco tienen impacto en la integridad de los datos.
Mencioné “aprobado” en la lista anterior. Revisión por el propietario del proceso y el propietario del sistema, absolutamente. Revisión por otras cuatro personas sin conocimiento de sistemas o procesos que solo querían su nombre en el documento, sin sentido. Mi registro personal en un proyecto CSV era un cliente que demandaba un total de nueve revisores y aprobadores. Si un revisor no va a leer el contenido o entender el contenido, ¿qué representa su firma: un autógrafo para la posteridad? ¿Una confirmación de que creen que el crítico anterior probablemente lo leyó? No ayuda….
PRUEBAS
Se pueden obtener grandes beneficios al aplicar el pensamiento crítico a las pruebas y, por lo tanto, no sorprende que la excelente instigación de la FDA de los enfoques de Computer Software Assurance (CSA) tenga un enfoque significativo en las pruebas.
Yo estimaría de forma conservadora que los scripts de prueba detallados click a click tardan cinco veces más en escribirse que en ejecutarse. La mayor proporción de defectos encontrados por dichos scripts de prueba son, sin duda, errores en los propios scripts. Veamos la motivación para scripts de prueba tan detallados.
Motivación Pensamiento Crítico
Debe desafiar una ruta o rama específica en el software; las instrucciones detalladas aseguran que llegue al camino o ramal. Esto aporta valor en la detección de defectos y beneficios finales para la seguridad del paciente.
El evaluador no sabe cómo operar el software, por lo que necesita las instrucciones de click a click.
Hoy quiero compartir con Uds, este artículo escrito por Thomas Peither, GMP-Verlag Peither AG

Un sistema CAPA debe entenderse como un elemento importante del sistema de calidad farmacéutica e implementarse de manera uniforme en toda la empresa o grupo. En principio, se puede implementar de dos formas diferentes: como un sistema autónomo o como un sistema integrado. En este artículo, profundizaré en los pros y los contras de cada uno, y también compartiré algunas de las mejores prácticas generales de CAPA.
- Sistemas CAPA Autónomos
Un sistema CAPA autónomo se caracteriza por su propia documentación y seguimiento de horarios. Un SOP autónomo describe el proceso para la definición, responsabilidad, seguimiento del cronograma y cierre de las actividades CAPA. Estas se derivan como posibles actividades de seguimiento a incidencias que están reguladas en otros sistemas, como por ejemplo para la atención de desviaciones, queja / reclamos o autoinspecciones.
En base a una queja, por ejemplo, se inicia un proceso CAPA en el sistema CAPA independiente después de su evaluación de riesgos, si es necesario, y se realiza un seguimiento mediante un sistema de documentación separado.
Los sistemas autónomos tienen una ventaja decisiva: se pueden tomar medidas inmediatas para eliminar la desviación y aún se pueden documentar y procesar correcciones rápidas dentro del sistema de activación, evitando así una generación inflacionaria de procesos CAPA.
Otra ventaja de un sistema autónomo es una clara separación entre el sistema CAPA y otros procesos. Por lo tanto, cada uno de los sistemas puede ser monitoreado, documentado y discutido por sí solo.
Sin embargo, esto también conlleva una desventaja: los dos procesos separados deben referenciarse entre sí para vincularlos de manera clara y comprensible. Esto siempre conlleva el riesgo de que la información se pierda o se transfiera incorrectamente.
Además, eventos similares que pueden no ser espectaculares cuando se ven individualmente pueden convertirse en debilidades del sistema cuando se ven como un todo, lo que es más difícil de reconocer cuando se ven por separado.
Los sistemas de IT son, por supuesto, útiles aquí. Cada uno genera procesos específicos para las diferentes actividades, pero están interrelacionados y hacen accesible toda la información del otro proceso.
- Sistemas CAPA Integrados
Un sistema CAPA integrado suele ser algo más simple. Esa es una clara ventaja. Aquí, al final del procesamiento del incidente inicial, se definen las medidas CAPA correspondientes en la misma hoja de documentación y posteriormente también se documenta su implementación. Después de manejar tanto el problema como las medidas CAPA, la unidad de calidad responsable puede evaluar y cerrar ambos procesos juntos.
Esta ventaja, por otro lado, hace que el proceso sea más complejo: una vez finalizado todo el proceso, se debe transferir de nuevo a la unidad de calidad para su evaluación estadística (análisis de tendencias, revisión por la dirección, etc.) y cierre en el sistema de gestión de calidad. Si hubiera inquietudes por parte de la unidad de calidad, el sistema debe otorgar a la unidad de calidad un derecho de rechazo. En este caso, el procedimiento será transferido nuevamente a la unidad responsable para lograr una conclusión aceptable.
Aspectos importantes para ambos sistemas CAPA
Autónomo o integrado, para ambas variantes, la descripción del sistema CAPA debe hacerse en el manual de gestión de la calidad junto con la descripción de los sistemas para desviaciones, quejas, retiros, etc.
Además, un sistema CAPA requiere su propio SOP en el que se definen los procedimientos junto con las responsabilidades y la documentación necesaria.
Tiene sentido prescribir un análisis de riesgo posterior a la investigación y procesamiento de las desviaciones y, en consecuencia, la decisión sobre el posible inicio de una actividad CAPA.
No importa a qué resultado se llegue, en todo caso se documenta que se ha pensado en una medida más en el sentido de CAPA.
Tres aspectos son importantes aquí:
- Seguimiento rápido de la implementación,
- Control de eficacia y
- Informar las actividades de CAPA en la revisión por la dirección.
¿Cuáles son las características de un sistema CAPA eficaz?
En última instancia, todos los sistemas CAPA efectivos se basan en los mismos procesos. Al definir las medidas CAPA, es importante determinar cómo se puede comprobar su eficacia. Para ello, se necesitan criterios objetivos que puedan utilizarse para determinar el resultado. Si la medida tomada no muestra el éxito esperado, se debe considerar una nueva acción correctiva o preventiva. Por lo tanto, un sistema CAPA es un proceso iterativo continuo que sigue hasta que conduce a un resultado satisfactorio.
Veamos un ejemplo: la primera acción es siempre la corrección y eliminación inmediatas de la desviación, seguida inmediatamente por una investigación de la causa del error. Una vez que se ha identificado la causa, se definen las acciones correctivas apropiadas para evitar que vuelva a ocurrir. Una vez que estas medidas hayan demostrado ser efectivas y sostenibles, este proceso estará completo.
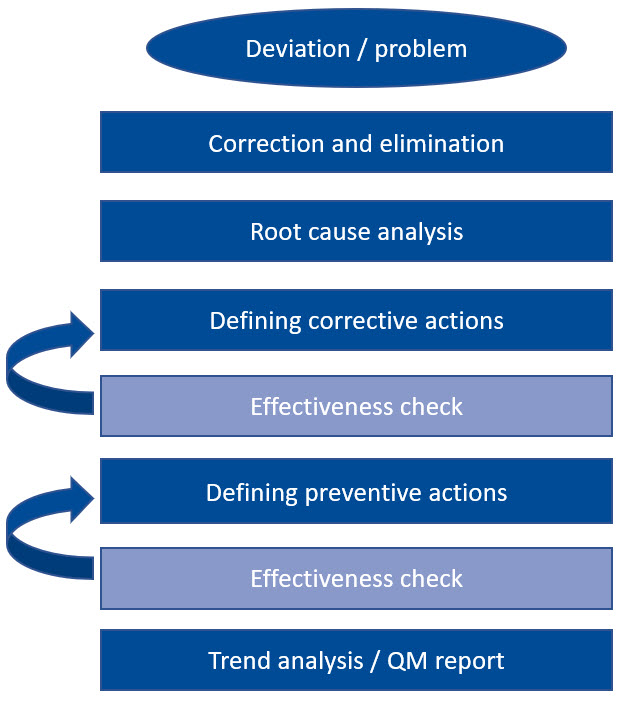
En principio, lo mismo se aplica a las acciones preventivas. Solo cuando se ha comprobado su eficacia se realiza un análisis de tendencias y todo se registra en el informe de gestión de calidad. Un enfoque tan rígido y pragmático no solo suena factible, sino que también es extremadamente práctico.
Hay varias razones por las que los sistemas CAPA, sin embargo, se han descarrilado con bastante facilidad en algún momento o todavía están en marcha. Por ejemplo, como suele ser el caso cuando se introduce un nuevo paradigma en la garantía de calidad farmacéutica, se observó un enorme aumento en las medidas CAPA en el período inicial cuando se establecieron los sistemas CAPA.
Esto se debió sólo en parte a una mayor conciencia de las interrelaciones pertinentes. Mucho más frecuentemente, se debió simplemente a malentendidos, como la evaluación de que toda no conformidad debe necesariamente resultar en una medida CAPA. Sin embargo, el Capítulo 1 de la Guía GMP de la UE no estipula la investigación de desviaciones o el inicio de medidas CAPA para cada no conformidad, sino que las limita a las desviaciones “relevantes”. Por lo tanto, es importante brindar a los empleados la capacitación suficiente y utilizar ejemplos para aumentar su conciencia sobre qué es una desviación y cómo debe documentarse.
Si una desviación documentada en el sitio se procesa más en el sistema CAPA o se puede corregir rápidamente en el sitio, lo decide una unidad superior o el control de calidad. En todo caso, deberá estar regulado en el SOP asociado.
Los involucrados en el proceso definitivamente deben participar en la evaluación de las no conformidades, porque conocen los procesos y las interrelaciones, tienen experiencia con las posibles causas y deben poder tomar decisiones rápidas.
La evaluación o clasificación rápida de un evento en particular no siempre tiene que ser realizada por un departamento de calidad superior. El experto en el sitio está, al menos según el entendimiento europeo de GMP, igualmente autorizado para ayudar a tomar decisiones. Como suele ser el caso, las mejores decisiones las toman todas las partes involucradas.
Es deseable, pero hasta ahora solo realizado en unas pocas empresas, vincular el instrumento de acción preventiva prospectiva con otros métodos de mejora continua de procesos como Six Sigma o Excelencia Operacional. Entonces, en lugar de usarse de forma rutinaria para cada desviación, en realidad solo podría aplicarse específicamente y con alta prioridad después de la evaluación de riesgos.
3 consejos para la vida diaria de GMP
Haga la vida un poco más fácil para los tomadores de decisiones y agregue una selección de medidas CAPA fijas al final del SOP para manejar las desviaciones, como cambios en las instrucciones de trabajo, cambios en los procesos, aumento en las frecuencias de prueba o monitoreo, cambio en las responsabilidades, o reentrenamiento de los empleados.
Use la medida estándar de “reentrenamiento de empleados” de uso frecuente con moderación. Una instrucción breve y directa a los empleados como medida correctiva suele ser más eficaz que una nueva formación formal.
Si la misma desviación o una similar ocurre una y otra vez, es hora de mirar más de cerca. Haga correcciones reales, tómese el tiempo para reescribir un SOP confuso y desordenado. Procesos, procedimientos, pautas y documentación más simples a menudo traen mejoras duraderas.
Este artículo se basa en el conocimiento de GMP contenido en el portal en línea GMP Compliance Adviser, que brinda información detallada sobre las mejores prácticas y regulaciones de GMP de Europa, EE. UU., Japón y muchos más (PIC/S, ICH, OMS, etc.). ).
Sobre el Autor:
Thomas Peither es miembro de la junta y director de marketing y desarrollo empresarial de GMP-Verlag Peither AG, Schopfheim, Alemania. Desde 2008, también dirige GMP Publishing Peither, Inc. Tiene más de 20 años de experiencia como consultor de GMP para empresas farmacéuticas. Se le puede contactar en thomas.peither@gmp-verlag.de.
Leyendo un artículo del Pharmaceutical on line, de Peter H. Calcott, D.Phil, tome unos párrafos de la misma que me parecieron sumamente importantes, por la claridad de conceptos sobre algunos temas que usualmente a todos nos generan dudas.
¿Qué escala debo usar en la Gestión de Riesgos de Calidad?
La gestión de riesgos de calidad (GRC) consiste en convertir opiniones en datos o números. Pero lidiar con esos números puede ser desalentador. ¿Qué escala debemos usar? ¿La escala de tres, cuatro, cinco o diez puntos? Tiendo a favorecer los tres o cinco, dependiendo de la situación.
Para un análisis preliminar de peligros (PHA), tiendo a favorecer una escala de tres puntos. Básicamente, es alto, medio o bajo. Es simple y fácil para tomar decisiones. En realidad, trata de no diferenciar la gravedad y la probabilidad entre sí. Simplemente puntúa el riesgo. Esto es útil para un análisis inicial o menor.
Para un análisis más detallado usando FMEA, prefiero una escala de cinco puntos. Da suficientes posibilidades sin ser demasiado complejo. A menudo, con una escala de 10 puntos, los miembros discuten entre un 7 y un 8. Eso simplemente no es importante. Incluso en la escala del 1 al 5, defina lo que cada uno representa para la gravedad y la probabilidad. No es necesario que sea demasiado elaborado, solo una descripción verbal clara. En la mayoría de los análisis FMEA, examinamos la gravedad (¿qué es lo peor que podría pasar?) y la probabilidad (¿qué probabilidad hay de que suceda?).
Estas son mis preferencias porque son simples. Puede usar una escala diferente, y eso también está bien.
La guía ICH Q9 también exige examinar la detectabilidad e incorporarla en los cálculos. Prefiero mantenerlo separado. Hago una evaluación, pero hago el análisis sin ella. Hago mi remediación basado en la severidad y la probabilidad. Si no puedo reducir el peligro lo suficiente, ahí es cuando entra en juego la detectabilidad. Luego, trabajo para que el problema sea detectable. Si no puedo resolverlo, al menos sé cuándo está ahí.
Recuerde, sus números son subjetivos
A medida que realiza los cálculos y la asignación de riesgos, recuerde que los números que asigna son subjetivos. Es la interpretación de sus opiniones, a menudo con pocos datos duros, pero a veces, tal vez solo datos blandos. Trátelos como tales y no se obsesione con los números reales. Estos números no deben tratarse como especificaciones, con asignación de suspenso y aprobación. El propósito de los números es realmente agrupar los elementos en tres cubos. Los primeros son los que deben ser subsanados. El segundo se puede dejar solo. Es decir, no se hace nada. El tercer cubo es para los elementos que se encuentran en el medio. Entonces, hay que preguntarse, ¿Lo arreglo? ¿Lo dejo? ¿O lo observo?
Mucha gente me pregunta: “¿Cómo estableces el estándar para decidir en cuál trabajarás y en cuál no?” Mi respuesta es mirar el análisis y tendrá una idea de dónde, en función de algunos de los elementos que ya sabe intuitivamente que necesitan reparación o no. La mayoría cae en los dos cubos anteriores y la decisión es clara. Es el tercer cubo con el que es más difícil lidiar. Después de hacer el análisis, revise los resultados. Haz la pregunta, ¿se siente bien? Está perfectamente bien decidir remediar algo que puntúa bajo en riesgo o descartar algo que puntúa alto. La clave es documentar sus decisiones, junto con las suposiciones y los datos que utilizó. Este informe se revisará más adelante y es posible que decida cambiar de opinión. La clave es documentar las razones para el futuro.
Al final del día, mire el resultado y pregúntese si se ve bien y tiene sentido. Si la respuesta es sí, ya está. Si no, llévelo a través de otro ciclo.
Elementos importantes a tener en cuenta
Entonces, ha completado el análisis. Eso significa que tiene las herramientas para ayudarlo a priorizar el trabajo y asignar los recursos adecuados para los problemas y las soluciones. Después de la remediación, es aconsejable volver a realizar el ejercicio para asegurarse de que las soluciones han reducido el peligro a un nivel de riesgo aceptable. Incluso entonces, no ha terminado.
Hay otras dos cosas con las que algunas personas tienen problemas. La primera es aceptar que el riesgo siempre estará ahí. Nunca llegarás al riesgo cero. O te volverás loco para llegar allí o te arruinarás. Tienes que aceptar que siempre habrá algún riesgo residual. Lo aceptamos en la vida real. ¿Por qué no aquí?
El segundo es la creencia de que los reguladores no aceptarán su decisión de no hacer nada. De hecho, este miedo se cuantificó en una encuesta que un colega y yo realizamos hace varios años en toda la industria. Un porcentaje considerable de encuestados no estaba convencido de que los representantes de las agencias aceptaran sus decisiones. Pero las agencias han dicho que utilizarán las técnicas. He desarrollado dispositivos, que incluyeron mucho análisis de riesgo en el desarrollo, y he visto a los reguladores aceptar mis análisis de riesgo durante las inspecciones.
Otro elemento, no relacionado con los puntos anteriores, estaba relacionado con una observación que se hizo en una empresa que permanecerá en el anonimato, por razones obvias. Esa empresa le presentó a un colega mío un análisis de riesgos que estaba muy bien hecho y bien documentado. Detalló un análisis de si liberarían un producto basado en un análisis de riesgo. ¡Hasta aquí todo bien! Pero el análisis de riesgo no estaba relacionado con el cumplimiento, la seguridad del paciente o la calidad del producto. Más bien, estaba relacionado con si serían atrapados durante una inspección u otra interacción regulatoria y las consecuencias. Mi colega rápidamente lo cortó de raíz, como dicen. El mensaje para llevar a casa aquí es que el análisis de riesgos nunca debe usarse para justificar algo que usted sabe intrínsecamente que no es correcto.
Espero que les resulte útil, a mi me pareció muy interesante el análisis de Peter H. Calcott, D.Phil.
A la pregunta de si es posible fabricar productos medicinales y no farmacéuticos en el mismo equipo, la FDA da una respuesta clara en una Warning Letter actual: No, esto no cumple con las cGMP.
Como consecuencia de la fabricación de productos medicinales y no farmacéuticos en el mismo equipo, la FDA solicita una evaluación de riesgos para todos los medicamentos fabricados en este equipo. Se debe evaluar la posible contaminación por compartir el equipo. Además, se requieren planes para abordar la calidad del producto y el riesgo del paciente para cada producto en distribución (incluidos los posibles retiros del mercado – Recall).
La FDA también requirió planes sobre si la compañía tiene la intención de continuar fabricando productos medicinales y no farmacéuticos. Si ambos van a seguir fabricándose, la FDA quiere ver planes que muestren que se utilizará equipo dedicado para las operaciones respectivas.
El programa de validación de limpieza debe mejorarse en términos de las condiciones del “worst case o peor de los casos”. Se debe tener en cuenta:
- Medicamentos con toxicidades más altas
- Medicamentos con niveles más altos de ingrediente activo
- Medicamentos con baja solubilidad en el reactivo de limpieza
- Medicamentos con propiedades que los hacen difíciles de purificar
- Muestrear los lugares que son más difíciles de limpiar
- Tiempo de espera antes de limpiar – Dirty Holding Time
Además, la FDA aún exige la especificación de los pasos necesarios que se deben tomar en el sistema de gestión de cambios antes de que se pueda introducir un nuevo equipo o producto de fabricación. También se solicita un plan CAPA basado en una revisión retrospectiva de las operaciones de limpieza con mejoras sugeridas que incluyen plazos para la implementación, así como una descripción de las mejoras en el programa de limpieza, incluidas las mejoras en la eficacia de la limpieza y la verificación continua de la limpieza.
Puede encontrar la Warning Letter completa dirigida a System Kosmetik Produktionsgesellschaft für kosmetische Gmbh en el sitio web de la FDA.
Publicado en la NL de la ECA GMP – 12/10/2022
Podría decir, sin mayor error, que muchas de las actividades que efectuamos, son requeridas por las Agencias, pero también quiero hacer foco en la importancia de las mismas, en el valor agregado que tienen, en los beneficios que aportan a nuestra operación.
La gestión de riesgos (GR), no es la excepción y es más que un simple requisito normativo con el que las empresas farmacéuticas tienen que cumplir. La GR es un proceso de mejora continua. Es una metodología mediante la cual identificamos, analizamos, evaluamos, mitigamos y monitoreamos el riesgo a lo largo del ciclo de vida y es de suma importancia que esté incorporado dentro de la cultura de la empresa.
No solo se trata de tener en la empresa dos personas especialistas en la GR, los operadores, los analistas deben entender los principios de la GR, la idea de ser más proactivos que reactivos, ya que la GR nos da el potencial de reducir la posibilidad de que ocurra un riesgo y su impacto potencial.
¿Qué es la cultura del riesgo y cómo la logramos? y en este punto, quiero dejarles dos preguntas:
¿Estamos trabajando en la identificación del riesgo? y cuando lo identificamos, ¿Nos asusta o lo ignoramos?
El comportamiento frente al riesgo cuando lo identificamos, es una parte fundamental del proceso.
Por supuesto que escondernos o no reconocer el riesgo, dejándo todo como está, no es una posición aceptable.
Una vez que el riesgo es comprendido, es esencial la exploración de mitigaciones para ese riesgo o la formación de una justificación de aceptación del riesgo.
Esos son los dos componentes principales que luego equivalen a construir una cultura de riesgo. Pero un aspecto crítico en este proceso es:
¿De qué forma la Dirección impulsa esta Cultura? ¿Existe una apertura a identificar y hablar sobre el riesgo? ¿O preferiría que siguiéramos adelante, sin pensar en los posibles problemas que podrían surgir?
Una vez que se identifican los riesgos pronosticados, la asignación de recursos donde se necesitan para mitigar esos riesgos también es un desafío.
Es usual la asignación de recursos donde ocurren problemas y debemos tomar acciones correctivas o preventivas, pero cuando se trata de un riesgo potencial, sobrecalificamos: ¿Cuánto dinero debemos invertir en esa acción sino sabemos si realmente va a suceder?
Debemos sentirnos seguros con nuestro proceso y aceptar que identificar el riesgo puede hacernos mejores. Enseñar a nuestros equipos a usar los datos y aprovechar su capacidad para minimizar el riesgo son factores para fomentar la cultura del riesgo.
La validación de limpieza es una actividad requerida dentro de la Industria Farmacéutica.
Desde las regulaciones y los estándares de la Industria, la validación de limpieza es reconocida como una actividad de suma importancia para establecer que la contaminación cruzada de un producto está controlada de manera de asegurar la calidad del producto y la seguridad del paciente.
Es una actividad continua dentro de las cGMP, la cual necesita una inversión significativa de recursos y de tiempo.
Si tomamos como una referencia la guía ISPE, Validación de limpieza para el siglo 21, uno de los objetivos que desarrolla es la aplicación del análisis de riesgo basado en la ciencia, en el proceso de validación de limpieza.
Análisis de riesgo de la limpieza
Riesgo de acuerdo a los principios de la ICHQ9 es función de la severidad del peligro y la probabilidad de ocurrencia.
Para los propósitos de la validación de limpieza, podemos definirlo como:
Riesgo = Fc (Severidad del residuo, Prob. aparición del residuo, Detectabilidad del residuo)
El nivel de esfuerzo, formalidad y documentación del proceso de QRM (Quality Risk Mangement) debe ser proporcional al nivel de riesgo del proceso de limpieza.
Análisis de Riesgo de la limpieza
El objetivo de la inspección es asegurar que las bases para todo límite están justificadas científicamente, por lo tanto los límites deberían ser determinados a partir del peligro que el residuo del proceso representa. Esto puede ser determinado desde la revisión de la toxicidad de los mismos residuos.
El ADE (límite de exposición diaria aceptable), puede ser usado como una medida de severidad del peligro de la sustancia, y a partir del mismo efectuamos el cálculo del MSC (Máxima cantidad arrastrada permitida segura).
Además debe ser considerado el peligro potencial aportado por el diseño del equipo, por ejemplo considerar que el equipo sea fácil de limpiar, inspeccionar y muestrear.
En cuanto a los agentes de limpieza, los mismos deben ser seleccionados en base a principios científicos y con conocimiento de los peligros que pueden representar. Es deseable que los mismos estén incluidos dentro de la lista GRAS (Generally Recognized As Safe), en caso que no se encuentren dentro de esta lista, puede ser usado el ADE del mismo para determinar su nivel de riesgo.
También se debe evaluar el peligro de una posible carga biológica remanente y la posibilidad de proliferación microbiológica durante o después de un proceso de limpieza y los peligros que esto representa. Por ejemplo, es necesario abordar los peligros que se presentan al mantener el equipo en estado sucio o limpio.
Exposición de la limpieza
Luego que el peligro de una sustancia ha sido identificado, a partir de su ADE se determina la MSC (máxima cantidad permitida segura), siguiendo las etapas para minimizar y evaluar los niveles posibles de exposición.
Antes de usar un SOP de limpieza, debe ser efectuado un análisis de riesgos (RA) por ejemplo utilizando FMEA. Se calcula el RPN (Risk Priority Number), a partir de la severidad del residuo, el nivel del residuo y la posibilidad de detectarlo. Cuando el RPN es alto, deben ser tomadas acciones de mitigación de forma de disminuir el riesgo.
Si el riesgo no puede ser reducido a nivel aceptable, el producto podría requerir equipos dedicados para su elaboración o el uso de elementos desechables.
Cuando el RA indica que la contaminación microbiológica es una preocupación, tales como equipamiento estéril, holding time de equipos, etc. Deben ser obtenidos y evaluados datos, para determinar el nivel de exposición. Caso contrario, los datos microbiológicos pueden no ser necesarios.
Detección de limpieza
La habilidad para detectar un residuo cuando está presente, es un factor importante en la reducción del riesgo.
Hay distintos métodos para la determinación de residuos, inspección visual, TOC, conductividad, HPLC, etc.
La inspección visual de determinados residuos, método usado para la verificación de la limpieza, apropiado para productos de bajo riesgo.
La conductividad, es otro método utilizado para la detección de presencia de productos de bajo riesgo. Es muy utilizada para la detección del punto final en los sistemas CIP (Clean In Place).
TOC, Carbono Orgánico Total, es una herramienta poderosa, simple y rápida, aunque inespecífica para la detección de residuos de limpieza.
HPLC, herramienta muy sensible, específica, para detectar residuos. Es de amplio uso para la detección de residuos de validación de limpieza. Es el método cuando los anteriores no pueden ser utilizados. usualmente se parte del método de valoración del activo y se lo adapta al nivel de trazas y luego se valida.
Debemos disponer de procedimientos de limpieza que nos aseguren procesos de limpieza de alta Capacidad y en el caso de sustancias de alta toxicidad, disponer de un monitoreo apropiado de los mismos.
La evaluación de los datos con respecto a la criticidad es esencial y, hasta cierto punto, no es una actividad trivial. Los datos deben evaluarse en particular con respecto a la seguridad del paciente y/o la calidad del producto.
El término “datos críticos” aparece en un solo lugar en las Directrices GMP de la UE:
“4.27 – Debería existir un sistema para indicar observaciones especiales y cualquier cambio en los datos críticos”.
El Anexo 11 de GMP de la UE también menciona datos críticos en un solo lugar.
“6. Chequeos de exactitud: para los datos críticos ingresados manualmente, debe haber una verificación adicional sobre la precisión de los datos”.
Esto proporciona una base legal para revisar la entrada de datos críticos. Por lo tanto, una segunda persona debe verificarlo. Una alternativa sería un sistema computarizado validado.
Ejemplo:
La temperatura se mide en un tanque de agitación. Esta medición es parte del informe de lote y es relevante para la liberación. La temperatura se lee manualmente y se documenta manualmente. En este caso, se requiere una segunda persona para verificar la exactitud de los datos (principio de doble verificación).
O
La temperatura se registra electrónicamente a través de un sistema computarizado y se guarda electrónicamente. En este caso, habría que validar el sistema informático.
Volviendo a la evaluación de la criticidad de los datos: ni la sección 4.27 ni el Anexo 11 (sección 6) proporcionan mucha información sobre qué datos son críticos y cuáles no. No existe una definición legal de datos críticos en el entorno GMP. Si uno observa las diversas directrices y estándares en el área GMP, se puede encontrar una definición de datos críticos en VDI/VDE 3516 Parte 5 sobre “Validación en el entorno GxP – Tipos de datos crudos (raw data)”:
“Datos críticos:
Datos que tienen un impacto potencial en la seguridad del paciente, la calidad del producto y la integridad de los datos”.
Es decisión de la empresa (fabricante de productos medicinales) definir los datos a incluir.
La pregunta crucial es sin duda: ¿Existen datos relevantes para GMP que no sean críticos?
Esto no puede responderse unívocamente a partir del contenido de las bases legales vigentes. Sin embargo, la respuesta es “No”. Sin embargo, existen diferencias en la criticidad de los datos. La Figura 1 muestra un ejemplo correspondiente. Una clasificación de tres niveles tiene sentido:

Todos los datos son relevantes desde el punto de vista GMP, sin embargo varía la criticidad.
La criticidad es alta, media o baja.
PI 041 BUENAS PRÁCTICAS PARA LA GESTIÓN E INTEGRIDAD DE DATOS EN ENTORNOS REGULADOS GMP/GDP proporciona información sobre datos críticos, lo que también demuestra su importancia.
Ejemplo:
“5.4 Criticidad de los datos: por ejemplo: para una tableta oral, los datos del ensayo API generalmente tienen un mayor impacto en la calidad y seguridad del producto que los datos de friabilidad de la tableta”.
El documento destaca la importancia de los datos en términos de su influencia en decisiones como la liberación de lotes.
“5.4 Criticidad de los datos
5.4.1 La decisión en la que influyen los datos puede diferir en importancia y el impacto de los datos en una decisión también puede variar. Los puntos a considerar con respecto a la criticidad de los datos incluyen:
¿En qué decisión influyen los datos?
Por ejemplo: cuando se toma una decisión de liberación de un lote, los datos que determinan el cumplimiento de los atributos críticos de calidad son normalmente de mayor importancia que los registros de limpieza del almacén”.
Tomado de la News Letter de la ECA – 06.02.2019
- Requerimientos basados en el producto / proceso (pensamiento basado en el producto / proceso)
- Risk Assessment basado en la seguridad del paciente
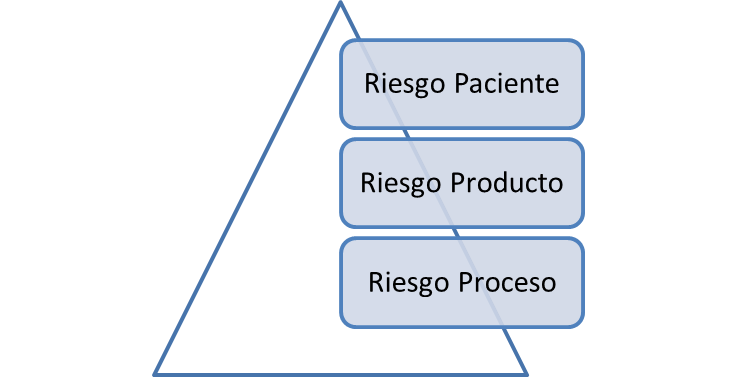
Cuando pensamos en Riesgo al producto, debemos considerar la seguridad, identidad, pureza, calidad y potencia del producto.
En cuanto a riesgos del proceso, debemos considerar los parámetros críticos del proceso, como temperaturas, tiempos de operación, presiones, IPCs, etc.
- Aumento del uso de las GEP (Buenas Prácticas de Ingeniería) – uso del FAT /SAT / Commissioning. Formas de trabajo de ingeniería.
- Testeos / ensayos de acuerdo al riesgo
- Reutilización de trabajos previos (ha sido hecho por otros, podemos confiar en otros) – know how, buenas prácticas
Según ISPE:
Requisitos: el URS debe basarse en el proceso y esto se confirma en el PQ (calificación de performance). Esto significa que tanto el IQ como el OQ están subordinados en importancia.
El diseño de experimentos (DOE) es un enfoque estadístico para la optimización de productos y procesos. Consiste en el estudio de distintas situaciones experimentales, que tienen sobre ciertas respuestas cuantitativas de unidades experimentales en observación.
De acuerdo a la definición de Wikipedia, el diseño experimental es una técnica estadística que permite identificar y cuantificar las causas de un efecto dentro de un estudio experimental. En un diseño experimental se manipulan deliberadamente una o más variables, vinculadas a las causas, para medir el efecto que tienen en otra variable de interés. El diseño experimental prescribe una serie de pautas relativas qué variables hay que manipular, de qué manera, cuántas veces hay que repetir el experimento y en qué orden para poder establecer con un grado de confianza predefinido la necesidad de una presunta relación de causa-efecto.
En la Industria farmacéutica actualmente es de suma importancia, constituyendo una parte sumamente importante del desarrollo de productos y la mejora continua del proceso.
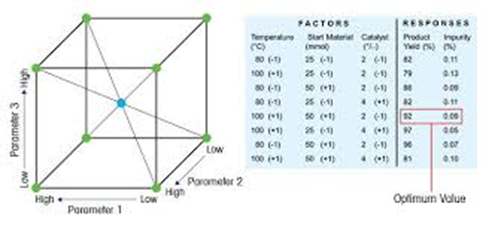
¿Qué ventaja aporta el DOE en el desarrollo farmacéutico?
Nos da la posibilidad de evaluar simultáneamente uno o varios parámetros de entrada, o “factores”, como la temperatura, la materia prima o la concentración, para determinar las condiciones en las que los atributos del producto, o “respuestas”, como el rendimiento, la selectividad o el nivel de impurezas, alcanzan un valor óptimo.
Eventos o pasos del DOE
Todo experimento involucra una serie de eventos:
1.Conjetura: hipótesis original que motiva el experimento ej. Marca de nafta vs rendimiento del coche.
2.Experimento: prueba efectuada para mostrar la conjetura.
3.Análisis: análisis estadístico de los datos obtenidos del experimento.
4.Conclusión: lo que se ha aprendido de la conjetura original con relación al experimento. A menudo conduce a una conjetura nueva.
En un próximo artículo daremos un ejemplo práctico del uso del DOE.
La capacidad del proceso (CP) es simplemente la relación entre la dispersión de los datos del proceso (su variabilidad) y el rango de las especificaciones de ese proceso. Básicamente, es una medida de qué tan bien puede encajar la dispersión de los datos dentro de su rango de especificación. Se dice que un proceso es capaz cuando la extensión de sus datos está contenida dentro de su extensión de especificación. Cuanto menor es la dispersión de los datos del proceso que la dispersión de la especificación, más capaz es un proceso.
Unos de los supuestos para estimar la capacidad del proceso, son que estos cálculos se efectúen con datos que se distribuyan normalmente y que el proceso sea estable (ausencia de tendencias significativas).
Capacidad de proceso de limpieza
Como hemos visto, la capacidad del proceso es una medida importante del desempeño del proceso que es relativamente simple de calcular, ya que solo requiere la media y la desviación estándar de los datos del proceso y los límites de especificación para ese proceso. Por lo tanto, en el caso de un proceso de limpieza, la capacidad del proceso de limpieza se puede calcular a partir de la media y la desviación estándar de los datos de hisopado o enjuague del proceso de limpieza y los límites de limpieza basados en el límite de exposición basado en la salud (HBEL) para los datos de hisopo o enjuague.
Para determinar la capacidad del proceso (CpU) de un proceso de limpieza, los términos de esta ecuación se pueden sustituir con los valores estimados a partir de los datos de limpieza (hisopo o enjuague) y el límite de limpieza basado en HBEL, como se muestra en la ecuación siguiente:
CpUlimpieza = (Límite de limpieza HBEL – Media datos de limpieza) / 3 S datos limpieza
Además, dado que los valores de capacidad del proceso se derivan de la media y la desviación estándar, se pueden calcular los intervalos de confianza superior e inferior para estos índices de capacidad del proceso. En los casos en los que puede haber tamaños de muestra variados o pequeños (p. ej., donde N suele ser < 25), es prudente y se recomienda informar y utilizar el límite de confianza inferior de la CPU de estos cálculos en lugar de solo la propia CPU.
Los softwares estadísticos como por ej. Minitab proporcionan estos valores de ntervalos de confianza para los valores de capacidad del proceso y también puede informar la cantidad esperada de fallas de un millón según el análisis de capacidad del proceso.
Capacidad de proceso de datos no normales
Como dijimos antes, partimos de la suposición de que los datos del proceso tienen una distribución normal, sin embargo no todos los datos de limpieza siguen una distribución normal. Por ejemplo, mientras que los datos de hisopos para el análisis de carbono orgánico total con frecuencia se distribuyen normalmente, los datos de HPLC con frecuencia no lo hacen. Antes de realizar cualquier análisis estadístico de los datos de limpieza, es importante determinar si los datos se distribuyen normalmente o no. Si los datos de limpieza no se distribuyen normalmente, se pueden utilizar distintas opciones para normalizarlos (ver el artículo: sus datos siguen una distribución Normal?).
Conclusión
Cada vez son más las compañías farmacéuticas que trabajan y entienden la importancia de analizar la capacidad de sus procesos. Entienden que valores < 1 son poco deseables y buscar obtener valores > 1,33 (equivalente a 4 sigmas).
Cuando pensamos en valores capacidad buscamos alcanzar valores mucho más altos y alcanzables. Evaluar la capacidad del proceso, nos permite conocerlo y además estimar cual es el riesgo para el paciente debido al arrastre de residuos y a partir del mismo alinearnos con el segundo principio de la ICH Q9, el nivel de esfuerzo debería ser proporcional al nivel de riesgo.