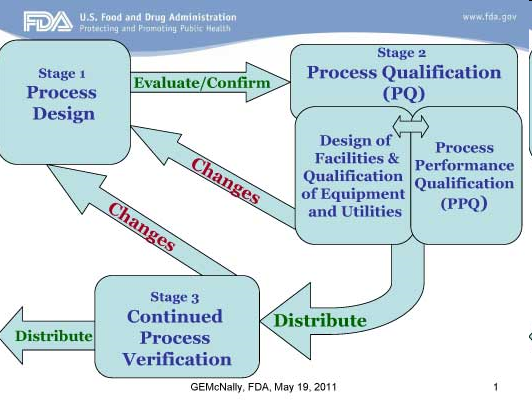
La calidad de los productos es un objetivo inherente de las compañías farmacéuticas. Es su misión desarrollar y producir productos, y entregar esos productos a los pacientes de forma segura. Sin embargo seguimos teniendo recalls, clausura de laboratorios, prohibiciones de productos, etc.
Quality by Design (QbD) ha impulsado a la industria a construir controles de calidad al inicio del ciclo de vida de un producto farmacéutico.
La FDA está buscando inputs de la industria sobre las métricas esenciales para el control de calidad para tomar decisiones basadas en el riesgo, quiere medidas objetivas de la performance de los sistemas de calidad del laboratorio, o sea busca ver métricas del producto y del laboratorio, para la comparación de tendencias a lo largo de la industria.
El uso de métricas parecería evidente para los gerentes de calidad y de Compliance, pero son un poco más resistidas por sus otros pares en general.
Podríamos pensar en cuales son los principales objetivos y beneficios del uso de métricas:
- Eliminar la subjetividad
- Proporcionar un punto de referencia y visibilidad para la mejora continua
- Asegurar el entrenamiento paralelo a través de múltiples funciones operativas, incluyendo garantía de calidad, operaciones y producción.

Métricas vs. Datos
Cuando pienso en este tema, recuerdo a E. Goldratt en el “Síndrome del Pajar” donde hace mención a que tenemos muchísimos datos, pero no siempre tenemos la información que necesitamos.
Aquí podríamos decir que tenemos cientos de miles de datos, a menudo desorganizados, inaccesibles e intrascendentes como herramientas de calidad.
Les dejo algunas métricas recomendadas para el laboratorio:
- % de Resultados OOS
- Tasa de Efectividad de CAPAs
- % Rechazo de lotes
- % de desvíos
- % de reclamos
- Capacidad de procesos (Cpk)
Curiosamente, estas métricas identificadas pueden generar luces rojas iniciales, tendencias que nos permiten preguntarnos:
¿Comenzó la desviación con la instalación de nuevos equipos o sistemas?
¿Se ha contratado un nuevo personal o se ha reducido la mano de obra existente?
¿Hay un nuevo gerente?
¿Se han cortado los presupuestos para el mantenimiento del equipo de producción?
También es interesante efectuarse estas u otras preguntas cuando las métricas muestran tendencias de mejora.
Sobre la base de esas respuestas, en el caso de tendencias negativas deberíamos encontrar las causas y generar planes de acción adecuados.
Las respuestas y acciones posteriores basadas en métricas útiles y precisas determinarán la calidad y el cumplimiento de una empresa.
Con el enfoque de la FDA sobre las métricas, la performance y la calidad, la forma en que una empresa recopila y usa métricas también identificará el riesgo de una empresa por fallas de calidad y si estos riesgos merecen mayor control e inspecciones por parte de la FDA.
Les dejo este par de preguntas para pensar:
¿Por qué tan pocas organizaciones integran con éxito las métricas en sus sistemas operativos?
y
¿Por qué seguimos viendo Recalls de productos muy comunes, cierres de plantas, acciones de cumplimiento y escasez de medicamentos?