Hoy quiero referirme a la Gestión de datos y hacer referencia a esta nota:
“No podemos evitar darnos cuenta de que la diferencia entre dato e información no reside en el contenido de un conjunto de caracteres dado. Más bien reside en su relación con la decisión requerida. Si no sabemos con antelación qué tipo de decisión vamos a tomar, si no sabemos qué vamos a necesitar exactamente, entonces todo dato podría llegar a considerarse como información en algún momento. ¿Resulta extraño que resulte tan difícil distinguir entre una base de datos y un sistema de información?… (Eli Goldratt, del capítulo 1 del Síndrome del Pajar).
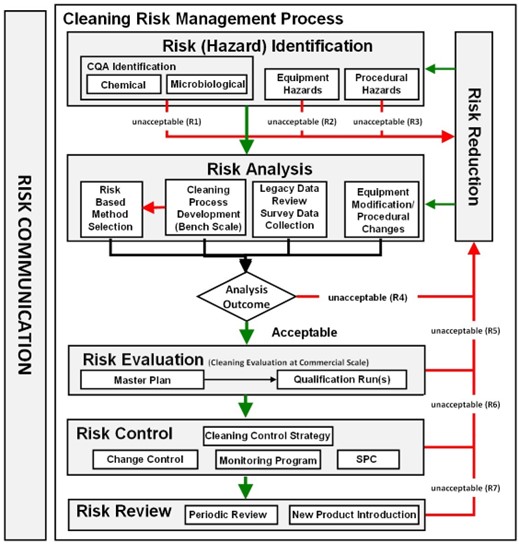
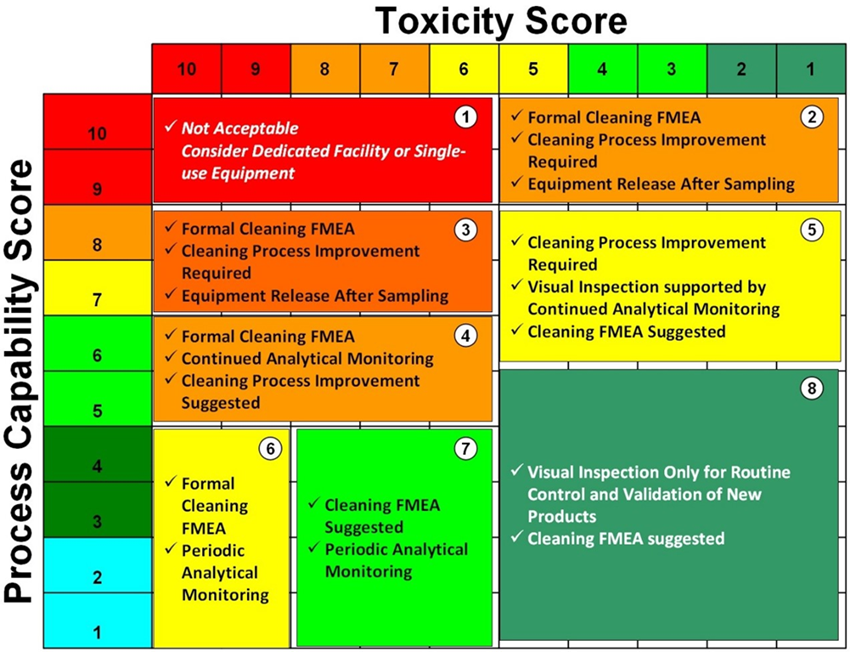
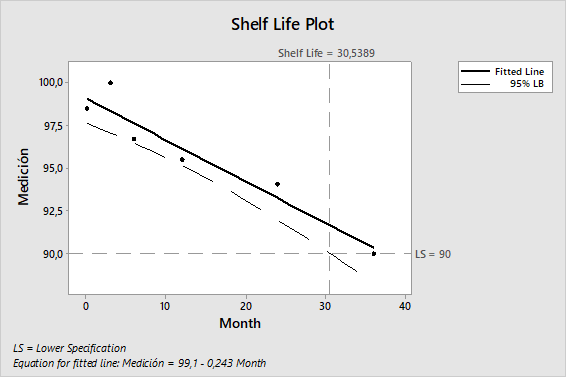
El propósito del análisis de tendencias es dar alertas tempranas de los atributos críticos de calidad o parámetros de proceso que muestran comportamientos no aleatorios que indican que el proceso está fuera de control estadístico y que pueden ser tomadas las acciones apropiadas para prevenir que los lotes estén fuera de especificación.
El monitoreo de los procesos a través de las tendencias es para mantener un estado de control y facilitar la mejora continua. Las tendencias deberían ser consideradas como una parte de la estrategia de control del proceso de manufactura.
Estos conceptos aplican a tendencia de datos de procesos y analíticos que son considerados críticos para la calidad de los productos elaborados por el laboratorio.
Por eso quiero proponerte, aprender trabajando, preparando a tu equipo de trabajo en el manejo tus datos, en el uso de herramientas estadísticas, que puedan obtener un valor agregado luego del enorme esfuerzo que significa efectuar las RAPs o la validación de los procesos.
Si está dentro de tus pendientes, comunicate con nosotros por info@cgmpdoc.com o por WA al 11 5008 2168, podemos coordinar una charla de 30 minutos y definir la forma de llevar adelante este tema,