A veces monitoreamos una medida a una frecuencia más baja de lo que es posible. Esto reduce la utilidad de la medida. La motivación para esto suele ser que ya tenemos reuniones semanales o mensuales para revisar las medidas y simplemente organizamos los datos en torno a esta frecuencia. Por lo tanto, controlamos los datos de los gráficos que se agregan a partir de un conjunto de datos más granular. Además, pensamos que las personas que miran los datos no estarán interesadas en los datos con mayor frecuencia. Sin embargo, cuando hacemos esto, podemos perder información significativa sobre el comportamiento actual y pasado de la medida que se está evaluando. Esto puede conducir a conclusiones incorrectas sobre lo que deberíamos estar haciendo con respecto a esta medida.
Como ejemplo, buscamos un conjunto de datos que representan el tiempo de proceso para un determinado producto. Los datos se promedian durante un período de tiempo mensual y se revisan en una reunión mensual. El gráfico de control de los datos durante un período de 12 meses se muestra en la figura 1.
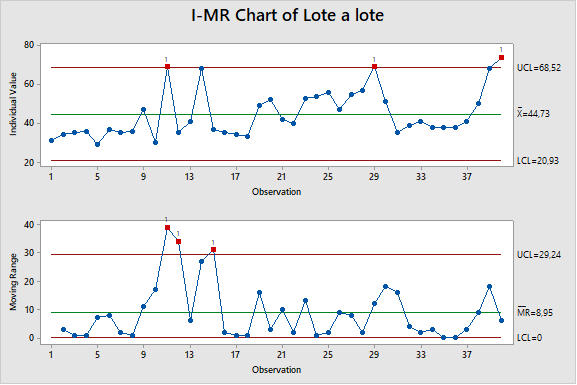
La figura 1 sugiere que podría haber una tendencia al alza, pero los datos no son concluyentes. Los datos no rompen ninguna regla de tendencia estadística, lo que no sorprende porque solo tenemos 12 puntos de datos. Se fabrican aproximadamente de 3 a 4 lotes de productos por mes, por lo que si observáramos los datos por lote, inmediatamente obtendríamos muchos más datos. El cuadro de control lote por lote se muestra en la figura 2.
Ahora tenemos 40 puntos de datos en lugar de 12. El comportamiento del tiempo de proceso ahora es mucho más claro.
Hemos tenido básicamente tres regiones de comportamiento durante el último año. Hay los siguientes:
Región 1: del lote 1 al lote 18. Tiene dos puntos de causa especial en el lote 11 y el lote 14 (en la gráfica solo hemos considerado la regla 1 de Shewhart). El promedio de los datos fue 39,0.
Región 2: del lote 19 al lote 30. No hay señales de causa especial. El promedio de los datos del período fue 52,1.
Región 3: del lote 31 al lote 40. Parece que las últimas 3 o 4 pérdidas de estos datos tienen una tendencia alcista.
Podemos ver en la figura 2 que la tendencia que sospechamos en la figura 1 se parece más a un cambio de paso en el lote 19. Por lo tanto, deberíamos preguntarnos qué cambió en el proceso entre los lotes 18 y 19. De manera similar, deberíamos preguntarnos qué cambió entre los lotes. 30 y 31 y lo que cambia entre los lotes 37 y 38. Ninguna de estas preguntas se haría si solo miramos la figura 1.
Por lo tanto, nos estamos perdiendo una gran cantidad de comportamiento del proceso al observar solo los datos promedio mensuales.
La lección aprendida es que no debemos agrupar datos al construir un gráfico de control. Se deben utilizar los datos más granulares disponibles.
Figura 1
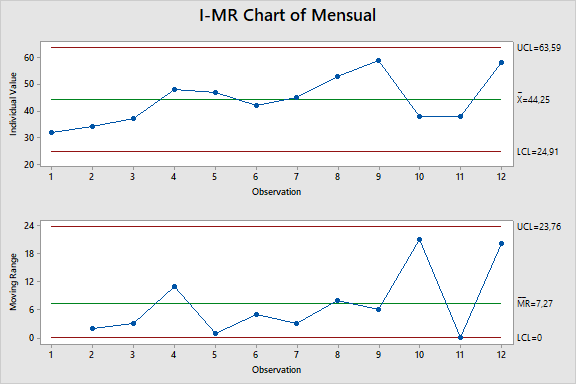
Figura 2
Este artículo fue adaptado del artículo de John McConnell, Brian Nummally y Bernard McGarvey publicado IVT.