El proceso de Análisis de riesgos conducido como parte del ciclo de vida de validación de sistemas computarizados, nos permite poner el énfasis y la atención apropiada en aquellas partes del sistema que pueden tener potencial impacto sobre la seguridad al paciente, la calidad del producto y la integridad de datos.

Es recomendable conformar un equipo para el manejo de la validación de los sistemas computarizados inicialmente, y para el posterior manejo de los cambios que pudiera tener el sistema durante su uso productivo. Este equipo debe tener como mínimo de los siguientes roles:
- Propietario del sistema (inicialmente este role puede ser llevado a cabo por el manager del proyecto)
- Propietario de procesos del negocio
- Proveedor (o representante técnico)
- Representante de IT (con formación en Compliance)
- Representante de Quality Assurance
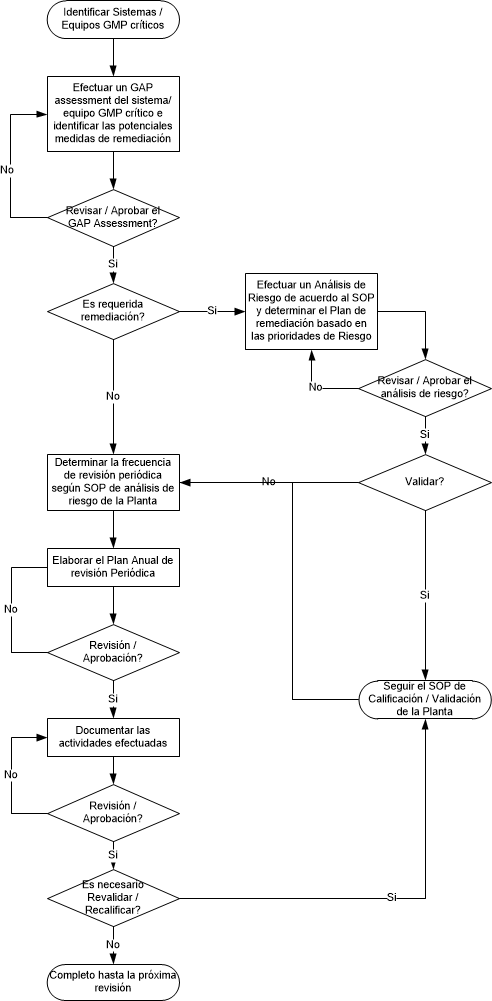
Los principios de evaluación y gestión de riesgos son aplicados desde que se introduce un sistema informático a la planta, y además durante el control de cambios, la revisión periódica y el decomiso del mismo.
Como parte de la implementación inicial del sistema computarizado, se evalúan los riesgos del proyecto, por ejemplo: restricciones de recursos, falta de conocimiento técnico, financiación, estabilidad del proveedor, etc. Estos riesgos del proyecto pueden ser manejados por el Propietario de procesos del negocio o Project Manager.
Registro del sistema y determinación del impacto regulatorio
Se trata de la primera etapa de análisis de riesgos del sistema, donde se efectúa una evaluación regulatoria (por ej. si el sistema tiene impacto GxP), o es crítico para el negocio, SOx, Finanzas, Privacidad de datos, eDiscovery. Luego de esta evaluación, se toma y se registra la decisión sobre si el sistema será validado.
Clasificación de riesgo del sistema
Si un sistema computarizado debe validarse entonces debe llevarse a cabo una clasificación de riesgo del sistema para ayudar a determinar el nivel y el alcance de las actividades de validación requeridas. Los resultados de la clasificación de riesgos deben ser registrados.
La clasificación de riesgos del sistema asigna un score que puede ser Alto, Medio o Bajo basado en:
- El status GxP del sistema
- El status de evaluación del proveedor (Rojo, Amarillo o Verde)
- La categoría GAMP5 del sistema
- Si el sistema almacena registros GxP
- Si el sistema tiene una red de conectividad.
Estos factores son registrados en el sistema de registro.
Esta clasificación de riesgo pretende reflejar el potencial nivel de riesgo que el sistema tiene para el proceso de negocio, por ej. Si el sistema fue desarrollado a medida, de acuerdo a las GAMP es categoría 5, o el proveedor tiene un sistema de gestión de calidad que NO es lo suficientemente maduro, la clasificación de riesgo del sistema establecida será mayor para asegurar que se aplican controles más exhaustivos durante el ejercicio de validación del sistema.
Para cada sistema, los entregables requeridos son descriptos en el Plan de Validación del sistema computarizado. El nivel de las actividades de validación dependerá de la clasificación del sistema, bajo, medio o alto, el paquete de testeos y su ejecución podría ser efectuado por el proveedor en el caso de un sistema de bajo riesgo, aunque en el caso de un sistema de alto riesgo, el paquete suele ser desarrollado en conjunto (proveedor + laboratorio) y la ejecución puede ser efectuada por el proveedor con la aprobación final del laboratorio o efectuada por el laboratorio.
Análisis de riesgo funcional
Si es requerido, un análisis de riesgo funcional es llevado a cabo. Esto se puede hacer a un nivel alto y / o un nivel funcional. El factor clave para llevar a cabo una evaluación de riesgos funcional es en los casos donde los escenarios de riesgo y la probabilidad y controles de mitigación no pueden determinarse sólo por hacer una evaluación de alto nivel y donde se requiere más detalle. Evaluación del riesgo sólo puede llevarse a cabo una vez que el diseño del sistema se ha acordado y aprobado. Los sistemas de bajo riesgo no necesitan someterse a una Evaluación de Riesgos. Los sistemas clasificados como de medio y alto riesgo deben someterse a una evaluación de riesgos en un nivel alto como mínimo. Una evaluación de riesgo alto nivel considera requisitos en grupos.
- La primera etapa es para clasificar los requerimientos. Dentro del URS, para cada requerimiento:
- Establecer el impacto GxP
- Establecer la criticidad para el negocio
- Establecer cualquier otra área sujeta a regulación, ej. SOx, Financiera, Privacidad de datos, eDiscovery.
- La segunda etapa implica la realización del análisis de riesgo para cada requerimiento
El foco de la evaluación está en la interacción del sistema con el proceso de negocio.
El análisis de riesgo debe considerar, para cada requerimiento:
- Qué podría salir mal (modos de fallas);
- Probabilidad
- Impacto
- Detectabilidad
Luego se lleva adelante un sistema de score de riesgos basados en la probabilidad de ocurrencia, el impacto o severidad del evento y la detectabilidad del mismo (según el enfoque de la GAMP5), el foco debe estar en aquellos ítems donde la clasificación final del riesgo del sistema es medio o alto.
A modo de ejemplo podemos utilizar los siguientes valores:
Probabilidad (1-3)
Severidad (1-3)
Detectabilidad (1-5)
De acuerdo a estos valores, el score mínimo es 1 y el máximo a obtener es 45 y podemos valorar los riesgos de la siguiente manera:
Bajo riesgo (1-4)
Riesgo medio (5-10)
Alto riesgo (>10)
- La tercera etapa en el proceso es para identificar los controles de mitigación para cada requerimiento. Los controles incluyen testeos técnicos y de negocios, SOPs, requerimientos de entrenamiento, etc.
- El reporte de la Evaluación de Riesgos funcional, puede ser elaborado utilizando un modelo o template adecuado, y el mismo debe ser aprobado por QA.