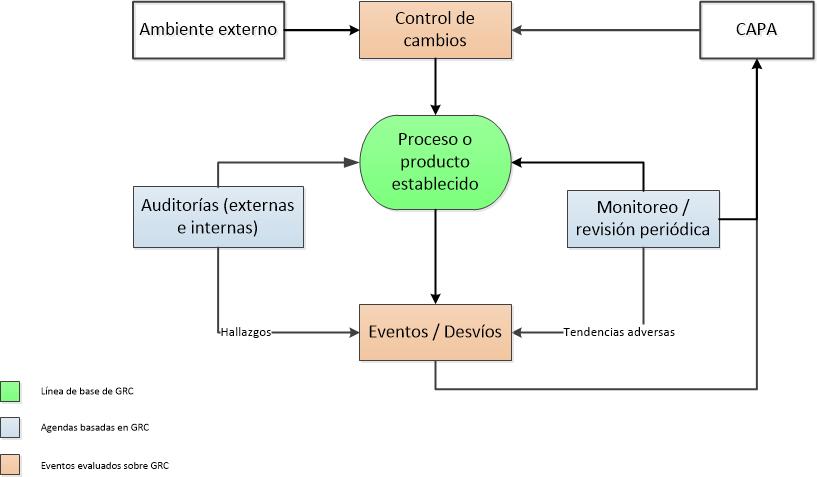
Hoy quiero referirme a un sistema de calidad, el sistema CAPA, sin embargo antes de comenzar a hablar específicamente sobre dicho sistema, quiero referirme al enfoque basado en el riesgo que están adoptando las agencias regulatorias y mencionar algunas preguntas que nos hacemos cuando intentamos comprender este tema, como por ejemplo: • ¿Cuál es el riesgo y para quién? • ¿Qué eventos causan o incrementan el nivel de riesgo? • ¿Cómo se manifiesta el riesgo? • ¿Cómo definir los niveles de riesgo? Pero antes veamos como la ICH Q9 define el riesgo:
“La combinación de la probabilidad de ocurrencia de daño y la gravedad de dicho daño. “Por lo tanto, el riesgo es asociado con un daño detectable, que puede ser medido a través de una probabilidad y severidad.
En el proceso de fabricación de productos, el riesgo se asocia con un evento que pueda comprometer la calidad, seguridad, y / o la eficacia del mismo. El medicamento comprometido podría dañar a los pacientes y al público en general, también en algunos casos, el riesgo podría afectar el personal de la fabricación de la droga, o a la empresa en sí, por ejemplo, ya que si la misma no cumple los requerimientos regulatorios puede ser multada o inhabilitada.
Centrándonos en el riesgo al paciente, es obligación del fabricante de medicamentos reducir la probabilidad de ocurrencia y minimizar la gravedad de los daños cuando estos eventos ocurren. Todas las actividades humanas y los emprendimientos tienen riesgo asociado con ellos. La elaboración de los medicamentos NO es la excepción.
Los eventos que suceden durante la elaboración de los medicamentos deben ser revisados y su riesgo evaluado.
Dependiendo del nivel del riesgo, la importancia o profundidad del análisis y la toma de acciones. Consideramos como de alto riesgo aquello que impacta directamente en el producto.
¿Cómo se manifiesta el Riesgo? Desde un punto de vista GMP, el riesgo depende del peligro que se plantea para el paciente cuando los eventos que suceden no son detectados y tratados adecuadamente antes de la la distribución del producto. Por ejemplo pueden ser liberados al mercado productos contaminados o con una potencia menor de la declarada.
Es sumamente importante ser consientes de las formas en que los riesgos se manifiestan, para poder tener un alto nivel de detectabilidad y tomar las acciones correctivas o preventivas apropiadas. Estos son componentes importantes para la estrategia de reducción de riesgos.
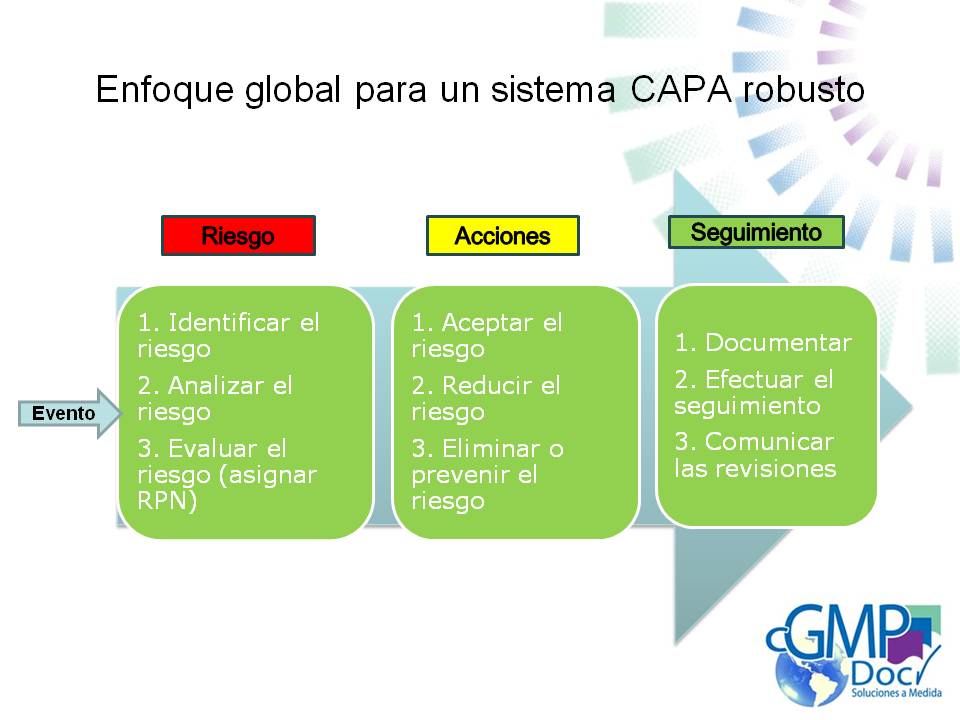
En orden de desarrollar la CAPA adecuada para mitigar el riesgo, uno tiene que definir y priorizar los niveles de riesgo.
Los niveles de riesgo y el Número Probabilidad de Riesgo (RPN) En la fabricación de productos farmacéuticos, el nivel de riesgo para una evento de calidad puede ser identificado a través de la combinación de la Severidad del daño al paciente, la frecuencia por la que la evento ocurre, y la detectabilidad del evento. Estos tres factores generalmente se les asigna un valor numérico y la multiplicación de los mismos da lugar al RPN= número de probabilidad de riesgo.
La severidad de un evento de calidad dado es una medida de la consecuencia del suceso en sí mismo y su potencial daño a la paciente, por ejemplo los valores más altos se lea signan a lesiones muy graves o muerte del paciente y los valores más bajos a eventos que no causan molestias. La frecuencia de un evento dado define la probabilidad de su aparición / reaparición. Los valores más altos son para una certeza que el evento ocurre u ocurrirá a situaciones que no pasan, quizás pasaron en el pasado, pero no ahora. La detectabilidad es una medida de la probabilidad de que la calidad evento se detecta o su efecto / resultado será fácilmente medido o visto. Aquí, los eventos que no son detectables tienen el índice de detectabilidad más alto, mientras que los eventos fácilmente detectables tienen el índice más bajo de detectabilidad.
Una vez que se determina el nivel de riesgo de un evento de calidad, es necesario aplicar los principios de Q9, donde indica que los esfuerzos de investigación y las acciones tomadas deben ser adecuados con la magnitud del problema y proporcionales a los riesgos encontrados.
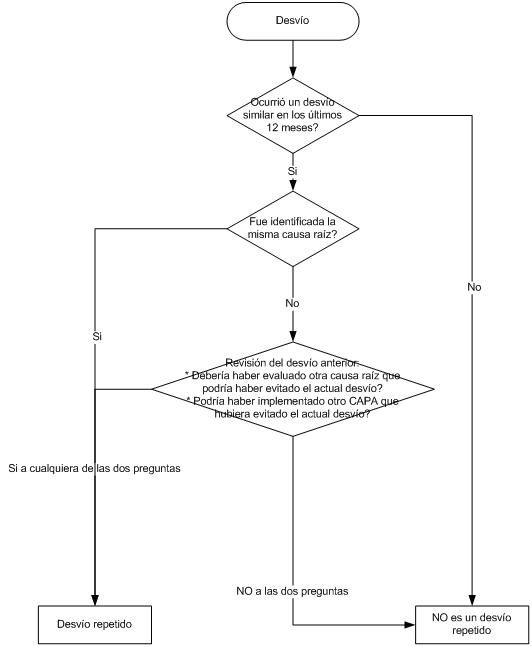
O sea que los eventos de alto riesgo requieren una investigación profunda, análisis de causa raíz, para luego tomar las acciones correctivas / preventivas apropiadas, ya que este evento NO debería volver a ocurrir.
CAPA (acciones correctivas y acciones preventivas) es un sistema de calidad diseñado para mitigar el riesgo en la elaboración de productos.
La ICHQ10 sugiere que las empresas farmacéuticas “Deben tener un sistema parala aplicación deacciones correctivas y preventivas… un enfoque estructurado paraelproceso de investigacióndebe ser utilizado conel objetivo dedeterminar la causa raíz”.
Como un sistema de calidad de reducción del riesgo, el sistema CAPA aborda eventos de calidad, que se producen durante el proceso de elaboración de productos para la salud. Como por ejemplo, desvíos o resultados OOS (fuera de especificación), etc.:
Estos eventos de calidad tienen el potencial de presentar riesgos para la población, de ahí la necesidad de mitigar su efecto.
Y como no podría ser de otra forma, el evento debe ser documentado, su investigación, las conclusiones, las acciones que deben adoptarse y el calendario para su aplicación, el cierre de la desviación o no conformidad y la verificación de la efectividad de la misma.
El seguimiento manual representa retos asociados con la generación de demasiado papel, siendo engorroso y consume mucho tiempo, además de proporcionar acceso limitado. El seguimiento electrónico se está convirtiendo en una alternativa cada vez más utilizada, ya que elimina muchas de las deficiencias del seguimiento de manual. Sin embargo, estos sistemas de rastreo electrónicos agregan el requisito de ser 21 CFR parte 11 compliance, ya que deben generar y mantener los registros electrónicos.
Las etapas del Sistema CAPA:

Hasta este punto, la discusión se ha centrado en el riesgo en cumplimiento y de una forma a grandes rasgos, el enfoque general de un programa de CAPA. Ahora veamos un poco más de cerca el sistema CAPA.
Como se mencionó anteriormente, CAPA es un sistema de aseguramiento de la calidad, que se ocupa de eventos de calidad, que se puede producir o podría ser previsto que se produzca durante la fabricación de productos para la salud. El sistema se basa en la revisión del evento y analizar el riesgo asociado con el mismo. Dependiendo del RPN, la necesidad de generar acciones de mitigación. Una vez tomada la decisión, entonces se toma la acción apropiada. El evento, el análisis, las decisiones tomadas, y la acción (s) tomada luego son documentados, comunicados y seguimiento a asegurarse de que eran correctos, adecuados, y no lo hicieron introducir variabilidad riesgo / diferente o adicional a la operación. El proceso en 10 pasos básicos:
- Evento
- Identificar y segregar el producto afectado (colocarlo ON HOLD)
- Identificar si hay un equipo involucrado (colocarlo ON HOLD)
- Documentar, iniciar el desvío y documentar las acciones inmediatas
- Investigar y evaluar el riesgo
- Identificación de la causa raíz
- Toma de las acciones correctivas necesarias para disminuir o eliminar el riesgo (asegurar que ningún cambio adicional introduce un nuevo riesgo)
- Registrar y comunicar
- Monitorear el proceso de ejecución
- Verificar la eficacia de la acción tomada
Resumen
Los eventos de calidad, que se producen durante la fabricación de productos de salud, siempre están asociados con un nivel de riesgo. Un programa CAPA robusto es un requisito reglamentario que define el nivel de riesgo y cómo mitigarlo. Sin embargo, debe tenerse en cuenta que la implementación de un programa de este tipo no solo se limita a cumplir con la norma, sino también favorece al negocio y el aspecto financiero de la compañía. . Por otra parte, la implementación de un programa de CAPA no sólo tiene un impacto económico positivo en la fabricación de tales procesos, sino que también daría lugar a una mejor satisfacción del cliente y reduce el riesgo para el público, el principal objetivo.