BRR (SOP y check list)
Si bien este SOP no necesita ser muy largo, necesita enfatizar la importancia de la exactitud a través de la revisión de los BR.
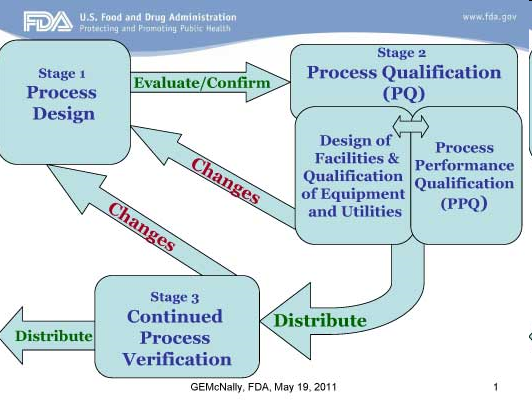
El SOP debería identificar las etapas claves del proceso de revisión. Esto incluirá pero no está limitado a lo siguiente:
- Verificación que todas las páginas del BR y de cualquier suplementos o adjuntos requeridos están presentes
- Verificación de la cronología de los eventos, procesos, tiempos de espera, evidentes y que están dentro de los rangos indicados
- Parámetros críticos del proceso, especificados para el producto, deberían ser revisados para asegurar que por ej. Tiempos, temperaturas, presiones, etc. están dentro de especificaciones
- El rendimiento de cada etapa en el proceso ha sido exactamente calculado y está dentro de especificaciones
- Las Buenas Prácticas de Documentación han sido seguidas, verificando los ingresos y las correcciones sobre el BR o cualquier adjunto al mismo
El SOP usualmente declara un tiempo para la revisión del BR incluyendo la necesidad de correcciones. Algunos laboratorios permiten 30 días desde que el registro del lote es recibido en QA. Posibles extensiones pueden ser permitidas si un desvío es citado y debe ser investigado. De todos modos las investigaciones de los desvíos tienen además que ser efectuadas en un tiempo razonable el cual asegurará que los BR sean cerrados oportunamente.
La revisión del BR debería ser más que un manejo de papeles robótico o un proceso puramente mecánico de buscar información incorrecta o faltante. Es importante, el BR nos da la historia completa del proceso de elaboración del lote.
El SOP de revisión de BR puede específicamente hacer referencia a una política o estándar u otro SOP que defina requerimientos de Buenas prácticas de documentación en el laboratorio.
Estos requerimientos típicamente incluyen, pero no están limitados a lo siguiente:
- Los ingresos deben ser legibles
- Los ingresos deben ser efectuados con tinta indeleble (preferentemente azul, aunque en algunos lugares la tinta negra es aceptada)
- Los ingresos deben ser fechados
- Los tiempos de estos ingresos deben ser consistentes con los estándares internos (AM/PM o 24 hs)
- Los ingresos son típicamente inicializados por el operador o los operadores que efectúan (u observan) la etapa. Un log de los nombres o iniciales será de alto valor para los revisores, pero una mejor opción es tener una página de firmas de los participantes del BR
- Cálculos, pesadas de los materiales y adición de los materiales al proceso deben ser verificados. La inicial y fecha (y usualmente hora) de la persona que efectúa la etapa y las iniciales y fecha de la persona que observó la etapa son ingresados / registrados en el BR.
- Correcciones, si las hay, deben llevar iniciales y fechas de las correcciones, en algunos casos es requerida una explicación de la corrección, para otros laboratorio es una recomendación
- Adjuntos listados, deben estar presentes y ser legibles
Este SOP debería proveer detalles sobre cuándo y cómo un revisor debería informar aquellos que son responsables de iniciar e investigar un desvío. Si hay información ausente o si un parámetro, incluyendo el rendimiento, está fuera de especificación.
Check list de BRR
El check list del BRR resalta los puntos clave que la revisión del BR debería cubrir y que requiere el revisor para sus sesiones de revisión.
Un check list específico debería ser preparado para cada tipo de producto, dado que la manufactura de un comprimido, es diferente a la de una cápsula, de un líquidos orales, etc. varían en cuanto a procesos y parámetros.
Productos para administración parenteral típicamente requieren testeos ambientales exhaustivos y testeos microbiológicos para confirmar esterilidad.
El check list suplementa al SOP, NO lo reemplaza.
A modo ilustrativo, las siguientes etapas pueden ser halladas en un check list genérico BRR que algunas firmas utilizan para documentar la revisión de cada BR.
Se pueden crear check lists que son específicos a un BR o proceso. Mientras esto agrega la responsabilidad de crear y mantener check lists adicionales, la revisión es permitida por medio de un check list que identifica las etapas críticas y otros parámetros relevantes para la operación bajo revisión.
Las siguientes son las etapas típicamente haladas en un check list de revisión de BR:
- Ingresar el nombre del producto, su potencia (tamaño del PKG si aplica) y el # de lote sobre el check list.
- Efectuar el conteo de las páginas y verificar que todas las páginas que son parte integral del BR están presentes y en orden. Usualmente las páginas del BR están numeradas de la siguiente manera: “Página x de XX”. Mire además durante la revisión la presencia de cualquier página adicional como copias de logs o páginas agregadas basadas en necesidades citadas en el BR.
- Verifique que una persona adecuadamente autorizada emitirá el BR y que lleva una firma y la fecha de emisión. Cada laboratorio debe determinar si el check list para la revisión de BR, el cual debería ser un documento controlado, debe ser emitido por una persona autorizada de la unidad de calidad o si puede ser impreso cuando se lo necesita desde un archivo electrónico el cual tiene acceso controlado por medio de un password u otro tipo de permisos.
- Liberación sobre estándares de documentación establecidos. Verificar que todas las entradas y correcciones están conformes de acuerdo a las expectativas del laboratorio. Estos requerimientos básicos fueron citados anteriormente.
- Un listado de BOM (Bill Of Materials) indicará que componentes y que cantidades son esperadas para un lote particular, contra el cual el revisor comparará las entradas del BR.
- El rendimiento del producto en términos de unidades de dosis o de masa total, debería coincidir con el rendimiento esperado (o sea dentro de los limites). Estos límites son típicamente absolutos y un revisor debería ser instruido para citar cualquier valor que pueda caer fuera del rango especificado. Ver 21CFR 211.188 (b).
- El BR debe ser firmado por una persona autorizada, usualmente, quién es el responsable de las etapas de manufactura. El revisor debe ser capaz de determinar a partir de documentos internos aquellos que están autorizados para firmar BRs.
- Si cualquiera carta o dato generado en proceso, debe ser incluido como parte del BR, verificar su existencia (ej. carta ambiental de la sala, datos de compresión como fuerza de compresión y rpm, temperaturas leídas de las soluciones en elaboración, registros de limpieza, etc.).
- Si cualquier excepción, es anotada en la revisión del BR, inicializar la etapa para corrección o si amerita, verificar que una investigación apropiada ha sido iniciada.
- Si el BR es para PKG, verificar que haya muestra de las etiquetas y que coincide con la esperada. La conciliación de los rótulos debe ser efectuada. Los conteos de las etiquetas son más exactas que la pesada de las mismas y conversión luego a un número aproximado.
- Si aplica, el revisor debe indicar una planilla separada o por medio de una entrada electrónica que se han notado excepciones y clarificaciones están siendo buscadas dentro del reporte de investigación del laboratorio y proceso de investigación. Esto es de suma utilidad para poder darle seguimiento a indicadores de calidad, relacionados al proceso de BRR.
- El check list del BR debe ser firmado y fechado cuando es completado.